Enantioconvergent Catalysis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Improved Catalytic Performance of Carrier-Free Immobilized Lipase by Advanced Cross-Linked Enzyme Aggregates Technology
Improved Catalytic Performance of Carrier-Free Immobilized Lipase by Advanced Cross-Linked Enzyme Aggregates Technology Xia Jiaojiao Jiangsu University of Science and Technology: Jiangsu University Yan Yan Jiangsu University Bin Zou ( [email protected] ) Jiangsu University https://orcid.org/0000-0003-3816-8776 Adesanya Idowu Onyinye Jiangsu University Research Article Keywords: lipase, carrier-free, immobilization, ionic liquid, stability Posted Date: May 24th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-505612/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/20 Abstract The cross-linked enzyme aggregates (CLEAs) are one of the technologies that quickly immobilize the enzyme without a carrier. This carrier-free immobilization method has the advantages of simple operation, high reusability and low cost. In this study, ionic liquid with amino group (1-aminopropyl-3- methylimidazole bromideIL) was used as the novel functional surface molecule to modify industrialized lipase (Candida rugosa lipase, CRL). The enzymatic properties of the prepared CRL-FIL-CLEAs were investigated. The activity of CRL-FIL-CLEAs (5.51 U/mg protein) was 1.9 times higher than that of CRL- CLEAs without surface modication (2.86 U/mg protein). After incubation at 60℃ for 50 min, CRL-FIL- CLEAs still maintained 61% of its initial activity, while the value for CRL-CLEAs was only 22%. After repeated use for ve times, compared with the 22% residual activity of CRL-CLEAs, the value of CRL-FIL- CLEAs was 51%. Further kinetic analysis indicated that the Km values for CRL-FIL-CLEAs and CRL-CLEAs were 4.80 mM and 8.06 mM, respectively, which was inferred that the anity to substrate was increased after modication. -

Chirality and Enantiomers
16.11.2010 Bioanalytics Part 5 12.11.2010 Chirality and Enantiomers • Definition: Optical activity • Properties of enantiomers • Methods: – Polarimetry – Circular dichroism • Nomenclature • Separation of enantiomers • Determination of enantiomeric excess (ee) Determination of absolute configuration • Enantiomeric ratio (E‐value) 1 16.11.2010 Definitions Enantiomers: the two mirror images of a molecule Chirality: non‐superimposable mirror‐images •Depends on the symmetry of a molecule •Point‐symmetry: asymmetric C, Si, S, P‐atoms •Helical structures (protein α‐helix) Quarz crystals snail‐shell amino acids Properties of Enantiomers – Chemical identical – Identical UV, IR, NMR‐Spectra – Differences: • Absorption and refraction of circular polarized light is different – Polarimetry, CD‐spectroscopy • Interaction with other chiral molecules/surfaces is different – Separation of enantiomers on chiral columns (GC, HPLC) 2 16.11.2010 Chiral compounds show optical activity A polarimeter is a device which measures the angle of rotation by passing polarized light through an „optical active“ (chiral) substance. Interaction of light and matter If light enters matter, its intensity (amplitude), polarization, velocity, wavelength, etc. may alter. The two basic phenomena of the interaction of light and matter are absorption (or extinction) and a decrease in velocity. 3 16.11.2010 Interaction of light and matter Absorption means that the intensity (amplitude) of light decreases in matter because matter absorbs a part of the light. (Intensity is the square of amplitude.) Interaction of light and matter The decrease in velocity (i.e. the slowdown) of light in matter is caused by the fact that all materials (even materials that do not absorb light at all) have a refraction index, which means that the velocity of light is smaller in them than in vacuum. -

Lipase-Catalyzed Kinetic Resolution of Dimethyl and Dibutyl 1-Butyryloxy-1-Carboxymethylphosphonates
catalysts Article Lipase-Catalyzed Kinetic Resolution of Dimethyl and Dibutyl 1-Butyryloxy-1-carboxymethylphosphonates Paulina Majewska Laboratory of Biotechnology, Department of Biochemistry, Molecular Biology and Biotechnology, Faculty of Chemistry, Wrocław University of Science and Technology, Wybrzeze˙ Wyspia´nskiego27, 50-370 Wrocław, Poland; [email protected]; Tel.: +48-71-320-4614 Abstract: The main objective of this study is the enantioselective synthesis of carboxyhydroxyphos- phonates by lipase-catalyzed reactions. For this purpose, racemic dimethyl and dibutyl 1-butyryloxy- 1-carboxymethylphosphonates were synthesized and hydrolyzed, using a wide spectrum of commer- cially available lipases from different sources (e.g., fungi and bacteria). The best hydrolysis results of dimethyl 1-butyryloxy-1-carboxymethylphosphonate were obtained with the use of lipases from Candida rugosa, Candida antarctica, and Aspergillus niger, leading to optically active dimethyl 1-carboxy- 1-hydroxymethylphosphonate (58%–98% enantiomeric excess) with high enantiomeric ratio (reaching up to 126). However, in the case of hydrolysis of dibutyl 1-butyryloxy-1-carboxymethylphosphonate, the best results were obtained by lipases from Burkholderia cepacia and Termomyces lanuginosus, leading to optically active dibutyl 1-carboxy-1-hydroxymethylphosphonate (66%–68% enantiomeric excess) with moderate enantiomeric ratio (reaching up to 8.6). The absolute configuration of the products after biotransformation was also determined. In most cases, lipases hydrolyzed (R) enantiomers of both compounds. Keywords: biocatalysis; hydroxyphosphonates; lipolytic activity; determination of absolute configu- Citation: Majewska, P. Lipase- ration; enantiomers Catalyzed Kinetic Resolution of Dimethyl and Dibutyl 1-Butyryloxy-1- carboxymethylphosphonates. Catalysts 2021, 11, 956. https:// 1. Introduction doi.org/10.3390/catal11080956 Enantioselective biocatalysis has long been used as an alternative to traditional meth- ods for obtaining pure chemical isomers [1]. -

Characteristic Features and Biotechnological Applications of Cross-Linked Enzyme Aggregates (Cleas)
Appl Microbiol Biotechnol DOI 10.1007/s00253-011-3554-2 MINI-REVIEW Characteristic features and biotechnological applications of cross-linked enzyme aggregates (CLEAs) Roger A. Sheldon Received: 11 July 2011 /Revised: 8 August 2011 /Accepted: 13 August 2011 # The Author(s) 2011. This article is published with open access at Springerlink.com Abstract Cross-linked enzyme aggregates (CLEAs) have reactors (no specialized equipment is needed) under mild many economic and environmental benefits in the context conditions (ambient temperature and pressure, physiological of industrial biocatalysis. They are easily prepared from pH) in an environmentally acceptable solvent (water) using a crude enzyme extracts, and the costs of (often expensive) biocompatible and biodegradable catalyst (an enzyme) that is carriers are circumvented. They generally exhibit improved itself derived from renewable resources. storage and operational stability towards denaturation by Enzymatic reactions proceed with high regio- and stereo- heat, organic solvents, and autoproteolysis and are stable selectivity and generally without the need for functional group towards leaching in aqueous media. Furthermore, they have activation and protection and deprotection steps required in high catalyst productivities (kilograms product per kilogram traditional syntheses. Hence, generally speaking, enzymatic biocatalyst) and are easy to recover and recycle. Yet another processes generate less waste than conventional synthetic advantage derives from the possibility to co-immobilize two routes, are more energy efficient, and provide products in or more enzymes to provide CLEAs that are capable of higher purity. The application of modern protein engineering catalyzing multiple biotransformations, independently or in techniques, such as directed evolution, has enabled the sequence as catalytic cascade processes. -

Advances in Asymmetric Oxidative Kinetic Resolution of Racemic Secondary Alcohols Catalyzed by Chiral Mn(III) Salen Complexes
Received: 28 March 2017 Revised: 30 June 2017 Accepted: 12 August 2017 DOI: 10.1002/chir.22768 REVIEW ARTICLE Advances in asymmetric oxidative kinetic resolution of racemic secondary alcohols catalyzed by chiral Mn(III) salen complexes Irshad Ahmad1 | Shagufta1 | Abdul Rahman AlMallah2 1 Department of Mathematics and Natural Abstract Sciences, School of Arts and Sciences, American University of Ras Al Khaimah, Enantiomerically pure secondary alcohols are essential compounds in Ras Al Khaimah, UAE organic synthesis and are used as chiral auxiliaries and synthetic intermedi- 2 Department of Chemical and Petroleum ates in the pharmaceutical, agrochemical, and fine chemical industries. One Engineering, School of Engineering, American University of Ras Al Khaimah, of the attractive and practical approaches to achieving optically pure second- Ras Al Khaimah, UAE ary alcohols is oxidative kinetic resolution of racemic secondary alcohols using chiral Mn(III) salen complexes. In the last decade, several chiral Correspondence Irshad Ahmad and Shagufta, Department Mn(III) salen complexes have been reported with excellent enantioselectivity of Mathematics and Natural Sciences, and activity in the homogeneous and heterogeneous catalysis of the oxida- School of Arts and Sciences, American tive kinetic resolution of racemic secondary alcohols. This review article is University of Ras Al Khaimah, Ras Al Khaimah, UAE. an overview of the literature on the recent development of chiral Mn(III) Emails: [email protected]; salen complexes for oxidative kinetic resolution of racemic secondary alco- [email protected] hols. The catalytic activity of monomeric, dimeric, macrocyclic, polymeric, Funding information and silica/resin supported chiral Mn(III) salen complexes is discussed School of Graduate Studies and Research, in detail. -

Lipase Immobilized on Mcfs As Biocatalysts for Kinetic and Dynamic Kinetic Resolution of Sec-Alcohols
catalysts Article Lipase Immobilized on MCFs as Biocatalysts for Kinetic and Dynamic Kinetic Resolution of sec-Alcohols Dominika Stradomska 1, Monika Heba 2 , Aleksandra Czernek 2, Nikodem Ku´znik 2 , Danuta Gillner 2,3 , Katarzyna Maresz 4, Wojciech Pudło 1, Andrzej Jarz˛ebski 1,4 and Katarzyna Szyma ´nska 1,* 1 Department of Chemical Engineering and Process Design, Silesian University of Technology, 44-100 Gliwice, Poland; [email protected] (D.S.); [email protected] (W.P.); [email protected] (A.J.) 2 Department of Organic Chemistry, Bioorganic Chemistry and Biotechnology, Silesian University of Technology, 44-100 Gliwice, Poland; [email protected] (M.H.); [email protected] (A.C.); [email protected] (N.K.); [email protected] (D.G.) 3 Biotechnology Center, Silesian University of Technology, 44-100 Gliwice, Poland 4 Institute of Chemical Engineering, Polish Academy of Sciences, 44-100 Gliwice, Poland; [email protected] * Correspondence: [email protected] Abstract: Dynamic kinetic resolution (DKR) is one of the most attractive methods for enantioselective synthesis. In the reported studies, lipase B from Candida antarctica (CALB) immobilized on siliceous mesoporous cellular foams (MCF) functionalized with different hydrophobic groups, and two ruthenium complexes with substituted cyclopentadienyl ligands were investigated as catalysts for the chemoenzymatic DKR of (rac)-1-phenylethanol, using Novozym 435 as a benchmark biocatalyst. Citation: Stradomska, D.; Heba, M.; Studies on the (rac)-1-phenylethanol transesterification reaction showed that CALB supported on Czernek, A.; Ku´znik,N.; Gillner, D.; MCFs grafted with methyl groups is a promising biocatalyst and isopropenyl acetate is a preferable Maresz, K.; Pudło, W.; Jarz˛ebski,A.; acylation agent. -

Review- Alternatives for the Separation of Drug Enantiomers: Ibuprofen As a Model Compound
Brazilian Journal of Chemical ISSN 0104-6632 Printed in Brazil Engineering www.abeq.org.br/bjche Vol. 23, No. 03, pp. 291 - 300, July - September, 2006 REVIEW- ALTERNATIVES FOR THE SEPARATION OF DRUG ENANTIOMERS: IBUPROFEN AS A MODEL COMPOUND P. O. Carvalho1*, Q. B. Cass2, S. A. Calafatti3, F. J. Contesini1 and R. Bizaco1 1Faculdade de Farmácia, Universidade São Francisco, Phone: +(55) (11) 4034-8298, Av. São Francisco de Assis 218, CEP 12916-900, Bragança Paulista - SP, Brazil. 2Departamento de Química, Universidade Federal de São Carlos, Phone: +(55)(16) 260-8208, Cx. Postal 676, CEP 13565-905, São Carlos-SP, Brazil 3Unidade Integrada de Farmacologia e Gastroenterologia Clínica, Universidade São Francisco, 12916-900 Bragança Paulista-SP, Brazil. E-mail: [email protected] (Received: March 5, 2005 ; Accepted: June 12, 2006) Abstract - Given the inherent dangers associated with racemic pharmaceuticals, exhaustive studies of techniques designed to separate enantiomers have been conducted. This paper reports a brief review of the different physical, chemical, and enzymatic methods used for the enantiomeric separation of rac-ibuprofen. The possible contribution of each technique to the preparation of enantiomerically pure drugs is reviewed in the context of competitive approaches that depend on the scale of application, with special emphasis on the recent progress achieved in this particular area. Keywords: Separation of drug enantiomers; Enzimatic methods. INTRODUCTION important. Another factor that is often overlooked in small-scale experiments is the volume yield or There are two possible approaches to the productivity (kilograms of product per unit reactor preparation of optically active compounds by volume per unit time). -

Biocatalysis in Continuous-Flow Mode: a Case- Study in the Enzymatic Kinetic Resolution of Secondary Alcohols Via Acylation
Biocatalysis 2017; 3: 27–36 Communication Open Access Juliana Christina Thomas, Martha Daniela Burich, Pamela Taisline Bandeira, Alfredo Ricardo Marques de Oliveira, Leandro Piovan* Biocatalysis in continuous-flow mode: A case- study in the enzymatic kinetic resolution of secondary alcohols via acylation and deacylation reactions mediated by Novozym 435® DOI 10.1515/boca-2017-0003 Received December 4, 2016; accepted February 7, 2017 Abstract: Enzymatic kinetic resolution reactions are a well- established way to achieve optically active compounds. When enzymatic reactions are combined to continuous- flow methodologies, other benefits are added, including reproducibility, optimized energy use, minimized waste generation, among others. In this context, we herein report a case study involving lipase-mediated transesterification by acylation and deacylation reactions of secondary alcohols/esters in batch and continuous-flow modes. 1 Introduction Acylation reactions were performed with high values of Enzymatic kinetic resolution (EKR) is a well-established enantiomeric excess (72 up to >99%) and enantioselectivity approach in the preparation of optically active compounds. (E > 200) for both batch and continuous-flow modes. On Nowadays, enzyme-mediated transformations, such as the other hand, for deacylation reactions using n-butanol EKR by lipases, are present in the toolbox of synthetic as nucleophile, enatiomeric excess ranged between 38 to chemists [1,2]. Enzymes are exceptional catalysts >99% and E from 6 to >200 were observed for batch mode. presenting enormous stereoselectivity for a number of For deacylation reactions in continuous-flow mode, natural and non-natural substrates, which allows the results were disappointing, as in some cases, very low or synthesis of valuable chiral intermediates, building- no conversion was observed. -
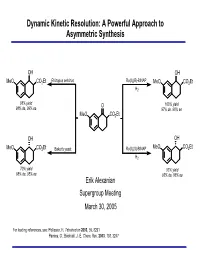
Dynamic Kinetic Resolution: a Powerful Approach to Asymmetric Synthesis
Dynamic Kinetic Resolution: A Powerful Approach to Asymmetric Synthesis OH OH Rhizopus arrhizus Ru(II)(R)-BINAP MeO CO2Et MeO CO2Et H2 98% yield O 100% yield 98% de, 99% ee 97% de, 96% ee MeO CO2Et OH OH MeO CO2Et Baker's yeast Ru(II)(S)-BINAP MeO CO2Et H2 70% yield 93% yield 98% de, 95% ee 93% de, 86% ee Erik Alexanian Supergroup Meeting March 30, 2005 For leading references, see: Pellissier, H. Tetrahedron 2003, 59, 8291 Pamies, O.; Backvall, J.-E. Chem. Rev. 2003, 103, 3247 Methods for Asymmetric Synthesis Synthesis utilizing existing stereogenic centers (chiral substrates or auxiliaries) Catalytic enantioselective organic reactions - Chemocatalysis - metal-mediated, Lewis acid-mediated, organocatalysis - Biocatalysis - enzymes (hydrolases) Resolution - Conventional separation procedures - Kinetic resolution kR SR PR kS E = kfast/kslow = 10 SS PS % ee 100 ee S Problems associated with standard kinetic resolutions: - Theoretical yield = 50% 50 ee P - Separation of product from remaining substrate required - In the majority of processes, only one stereoisomer is desired - Drop in enantiomeric purity as process nears 50% conversion 0 0 50 100 % conversion Dynamic Kinetic Resolution Classic Resolution Dynamic Resolution kR kR SR PR SR PR kinv kS kS SS PS SS PS kS E = kfast/kslow = 10 % ee 100 kR ∆∆G ee P DKR kinv 50 ee S ee P SS S 0 R 0 50 100 % conversion P PS R If racemization can occur concurrently with kinetic resolution, then theoretically 100% of the racemic mixture can be converted to one enantiomer. This process is known as dynamic kinetic resolution (DKR). DKR is an example of a Curtin-Hammett system in which the composition of products is controlled by the free energies of the transition states and not the composition of the starting materials. -

1 Introduction: a Survey of How and Why to Separate Enantiomers Matthew Todd
1 1 Introduction: A Survey of How and Why to Separate Enantiomers Matthew Todd This book is about the separation of enantiomers by synthetic methods, which is to say methods involving some chemical transformation as part of the separation process. We do not in this book cover chromatographic methods for the separation of enantiomers [1]. Nor do we focus on methods based on crystallizations as these have been amply reviewed elsewhere (see below). We are concerned mainly therefore with resolutions that involve a synthetic component, so mostly with the various flavours of kinetic resolutions through to more modern methods such as divergent reactions of a racemic mixture (DRRM). This introduction briefly clarifies the scope of the book. The reasons such methods are of continued importance are threefold: 1) Society: the need for enantiopure compounds. New molecules as single enan- tiomers are important to our continued well-being because they are the feedstocks of new medicines, agrochemicals, fragrances and other features of modern society in a chiral world. Of the 205 new molecular entities approved as drugs between 2001 and 2010, 63% were single enantiomers [2]. Nature provides an abundance of enantiopure compounds, but we seek, and need, to exceed this by obtaining useful unnatural molecules as single enantiomers, and we may reasonably want to access both enantiomers of some compounds. 2) Academia: the basic science involved in the behaviour of chiral compounds.If we seek the state of the art in our discipline, we cannot help but think that rapid and selective chemical distinction between enantiomers, which results in their facile separation, is something beautiful in itself. -

Enantiomeric Recognition and Separation by Chiral Nanoparticles
molecules Review Enantiomeric Recognition and Separation by Chiral Nanoparticles Ankur Gogoi 1,† , Nirmal Mazumder 2,†, Surajit Konwer 3, Harsh Ranawat 2 , Nai-Tzu Chen 4 and Guan-Yu Zhuo 4,5,* 1 Department of Physics, Jagannath Barooah College, Jorhat, Assam 785001, India; [email protected] 2 Department of Biophysics, School of Life Sciences, Manipal Academy of Higher Education, Manipal, Karnataka 576104, India; [email protected] (N.M.); [email protected] (H.R.) 3 Department of Chemistry, Dibrugarh University, Dibrugarh, Assam 786004, India; [email protected] 4 Institute of New Drug Development, China Medical University, No. 91, Hsueh-Shih Rd., Taichung 40402, Taiwan; [email protected] 5 Integrative Stem Cell Center, China Medical University Hospital, No. 2, Yude Rd., Taichung 40447, Taiwan * Correspondence: [email protected]; Tel.: +886-4-2205-3366 (ext. 6508); Fax: +886-4-2207-1507 † These authors contributed equally to this work. Academic Editors: Maria Elizabeth Tiritan, Madalena Pinto and Carla Sofia Garcia Fernandes Received: 3 February 2019; Accepted: 10 March 2019; Published: 13 March 2019 Abstract: Chiral molecules are stereoselective with regard to specific biological functions. Enantiomers differ considerably in their physiological reactions with the human body. Safeguarding the quality and safety of drugs requires an efficient analytical platform by which to selectively probe chiral compounds to ensure the extraction of single enantiomers. Asymmetric synthesis is a mature approach to the production of single enantiomers; however, it is poorly suited to mass production and allows for only specific enantioselective reactions. Furthermore, it is too expensive and time-consuming for the evaluation of therapeutic drugs in the early stages of development. -

Recent Trends in Whole Cell and Isolated Enzymes in Enantioselective Synthesis
Reviews and Accounts ARKIVOC 2012 (i) 321-382 Recent trends in whole cell and isolated enzymes in enantioselective synthesis Sinéad E. Milner a and Anita R. Maguire *b aDepartment of Chemistry, and bDepartment of Chemistry and School of Pharmacy, Analytical and Biological Chemistry Research Facility, University College Cork, Cork, Ireland E-mail: [email protected] Abstract Modern synthetic organic chemistry has experienced an enormous growth in biocatalytic methodologies; enzymatic transformations and whole cell bioconversions have become generally accepted synthetic tools for asymmetric synthesis. This review details an overview of the latest achievements in biocatalytic methodologies for the synthesis of enantiopure compounds with a particular focus on chemoenzymatic synthesis in non-aqueous media, immobilisation technology and dynamic kinetic resolution. Furthermore, recent advances in ketoreductase technology and their applications are also presented. Keywords: Biocatalysis, kinetic and dynamic kinetic resolution, Baker’s yeast, ketoreductases Table of Contents 1. General Introduction 2. Biocatalysis 2.1 Introduction 2.2. Enzyme classes 2.3. Advantages and disadvantages of biocatalysis 3. Biocatalysis in Non-Aqueous Media 3.1. Organic solvents 3.2. Supercritical fluids 3.3. Ionic liquids 3.4. Novel application of fluorous solvents in product isolation following biocatalysis 4. Enzyme Immobilisation 4.1. Introduction 4.2. Cross-linked enzyme aggregates 4.3. Practical applications of enzyme cross-linked enzyme aggregates 5. Genetic Engineering of Enzymes Page 321 ©ARKAT-USA, Inc. Reviews and Accounts ARKIVOC 2012 (i) 321-382 5.1. Rational design and directed evolution 5.2. Practical applications of genetically modified enzymes 6. Kinetic and Dynamic Kinetic Resolution – a Biological Perspective 6.1.