University of Southampton Research Repository
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

W B E Rlin Ie
K u p P a n Stanisław Sojka H istoria sportu WILNO p a ł a c (s. 2) W y w i a d (s. 3) ż u ż l o w e g o (s. 5) STYCZEŃ W spółczesny P a m i ę ć D z i e w i c a 1991 (s.2) D r a k u l a (s. 4) i d o k t o r (s. 6) (s. 7-8) SERWIS INFORMACYJNY „KRAJ-ŚW IAT” str. 7 - 8 Zielonogórska V Głogotuska Gorzowska Zainteresowanie Moskwą, Ro Zachód czeka i zgłasza swoją s ją i c a ły m z w ią z k ie m je s t o g r o m gotowość do natychmiastowego ne. Tym większe, im poważniej opanowania tego nąjwiększego i Kuszące dziew czyny szy staje się kryzys polityczny, jednocześnie najbardziej potrze gospodarczy i narodowościowy. bującego pomocy rynku świata. Obecność Zachodu staje się coraz Co oferujemy my? Praktycznie wyraźniejsza. Przed Mc Donal nie ma żadnych polskich towa dem nieustające kolejki. Na ulicy rów. Ze sklepów kosmetycznych Dymitrowa pojawiła się wielka zniknęła naw et Pollena czy Mira- plansza reklamowa Manhattan kulum, tak bardzo tu kiedyś po Bank. W największym domu to w B e r l i n i e pularne. Z jednej strony eksport warowym - słynnym GUM, no do ZSRR stał się m niej opłacalny, Z Zielonej Góry do Berlina jest stopnia, że zacząłem je fotografować, woroczne pozdrowienia od Ame- a z drugiej, nad naszymi stosun tylko 150 kilometrów. Spacerując Mój obiektyw utrwalał kolejne po Berlinie wieczorem mimo woli piękności. -

World Finals 1936-1994
No Rider Name 1 2 3 4 5 Tot BP TOT No Rider Name 1 2 3 4 5 Tot BP TOT No Rider Name 1 2 3 4 5 Tot BP TOT 1 Dicky Case 1 0 3 1 2 7 9 16 7 Ginger Lees 2 0 1 0 1 4 7 11 13 Bob Harrison 0 0 2 0 3 5 10 15 2 Frank Charles 3 3 0 2 0 8 12 20 8 Bluey Wilkinson 3 3 3 3 3 15 10 25 14 Eric Langton 3 3 3 2 2 13 13 26 3 Wal Phillips 1 1 0 2 1 5 7 12 9 Cordy Milne 2 2 1 3 3 11 9 20 15 Vic Huxley 1 2 0 2 2 7 10 17 4 George Newton 0 0 3 1 0 4 12 16 10 Bill Pritcher 0 1 0 0 1 2 6 8 16 Morian Hansen 2 1 2 0 0 5 10 15 5 Jack Ormston 1 1 2 3 1 8 9 17 11 Lionel Van Praag 3 3 3 2 3 14 12 26 R17 Norman Parker 1 1 6 7 6 Arthur Atkinson 0 2 1 0 0 3 6 9 12 Jack Milne 1 2 1 0 2 6 9 15 R18 Blazer Hansen 0 5 5 HEAT No. RIDER NAME COL COMMENTS No. RIDER NAME COL COMMENTS No. RIDER NAME COL COMMENTS PTS 1 Dicky Case R 2 2 Frank Charles R 0 2 Frank Charles R 0 2 Frank Charles B 3 6 Arthur Atkinson B 1 8 Bluey Wilkinson B 3 1 9 17 3 Wal Phillips W 1 11 Lionel Van Praag W 3 10 Bill Pritcher W 1 73.60 4 George Newton Y Fell 0 76.60 16 Morian Hansen Y 2 78.60 15 Vic Huxley Y 2 5 Jack Ormston R 1 1 Dicky Case R 3 1 Dicky Case R 2 6 Arthur Atkinson B 0 5 Jack Ormston B 2 7 Ginger Lees B 1 2 10 18 7 Ginger Lees W 2 12 Jack Milne W 1 9 Cordy Milne W 3 77.20 8 Bluey Wilkinson Y 3 78.60 15 Vic Huxley Y 0 78.80 16 Morian Hansen Y 0 9 Cordy Milne R 2 2 Frank Charles R 2 3 Wal Phillips R 1 10 Bill Pritcher B 0 7 Ginger Lees B 1 6 Arthur Atkinson B 0 3 11 19 11 Lionel Van Praag W 3 12 Jack Milne (US) W 0 12 Jack Milne W 2 75.80 12 Jack Milne Y 1 76.80 14 Eric Langton Y 3 79.80 13 -

CONTAMINANTS in the BALTIC SEA SEDIMENTS Results of the 1993 ICES/HELCOM Sediment Baseline Study
CONTAMINANTS IN THE BALTIC SEA SEDIMENTS Results of the 1993 ICES/HELCOM Sediment Baseline Study Matti Perttilä (Editor), Horst Albrecht, Rolf Carman, Arne Jensen, Merentutkimuslaitos Per Jonsson, Harri Kankaanpää, Birger Larsen, Mirja Leivuori, Havsforskningsinstitutet Lauri Niemistö, Finnish Institute of Szymon Uscinowicz & Boris Winterhalter Marine Research No. 50 2003 Report Series of the Finnish Institute of Marine Research MERI - Report Series of the Finnish Institute of Marine Research No. 50, 2003 CONTAMINANTS IN THE BALTIC SEA SEDIMENTS Results of the 1993 ICES/HELCOM Sediment Baseline Study Matti Perttilä (Editor), Horst Albrecht, Rolf Carman, Arne Jensen, Per Jonsson, Harri Kankaanpää, Birger Larsen, Mirja Leivuori, Lauri Niemistö, Szymon Uscinowicz & Boris Winterhalter MERI — Report Series of the Finnish Institute of Marine Research No. 50, 2003 Cover photo by Maija Huttunen Publisher: Julkaisija: Finnish Institute of Marine Research Merentutkimuslaitos P.O. Box 33 PL 33 FIN-00931 Helsinki, Finland 00931 Helsinki Tel: + 358 9 613941 Puh: 09-613941 Fax: + 358 9 61394 494 Telekopio: 09-61394 494 e-mail: surname@fimr fl e-mail: [email protected] Copies of this Report Series may be obtained from the library of the Finnish Institute of Marine Research. Tämän raporttisarjan numeroita voi tilata Merentutkimuslaitoksen kirjastosta. ISSN 1238-5328 ISBN 951-53-2557-9 Dark Oy, Vantaa 2003 CONTENTS ABSTRACT 5 1. INTRODUCTION 6 Matti Perttilä, Birger Larsen, Lauri Niemistö & Boris Winterhalter 2. THE 1993 HELCOM/ICES BALTIC SEA SEDIMENT BASELINE STUDY 9 Matti Perttilä, Per Jonsson, Birger Larsen, Lauri Niemistö, Boris Winterhalter & Walter Axelsson 3. MINERALOGICAL COMPOSITION AND GRANULOMETRY 21 Szymon Uzcinowicz, Wanda Narkiewicz & Krzysztof Sokowski 4. DISTRIBUTION OF TRACE METALS IN THE BALTIC SEA SEDIMENTS 25 Horst Albrecht, Matti Perttilä & Mirja Leivuori 5. -
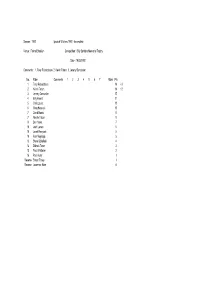
1992 Ipswich Witches 1992 - Incomplete
Season : 1992 Ipswich Witches 1992 - Incomplete Venue : Foxhall Stadium Competition : Billy Sanders Memorial Trophy Date : 19/03/1992 Comments : 1. Tony Rickardsson 2. Kelvin Tatum 3. Jeremy Doncaster No. Rider Comments 1 2 3 4 5 6 7 Rides Pts 1 Tony Rickardsson 14 +3 2 Kelvin Tatum 14 +2 3 Jeremy Doncaster 12 4 Billy Hamill 11 5 Chris Louis 10 5 Greg Hancock 10 7 David Norris 8 7 Neville Tatum 8 9 Ben Howe 7 10 Josh Larsen 5 10 Jacek Rempala 5 10 Alan Mogridge 5 13 Steve Schofield 4 14 Zdenek Tesar 3 15 Paul Whittaker 2 16 Paul Hurry 1 Reserve Shaun Tacey 1 Reserve Lawrence Hare 0 Season : 1992 Ipswich Witches 1992 - Incomplete Venue : Saddlebow Road Competition : Challenge (Anglian Cup) King's Lynn 46 Ipswich 44 Date : 21/03/1992 Comments : Home Team No. Rider Comments 1 2 3 4 5 6 7 Rides Pts BP FM PM 1 Bohumil Brhel 3 2 0 2 0 5 7 2 Dennis Lofqvist 1 1 1 2 4 5 1 3 Mark Loram 2 2 R 1 4 5 4 Gary Allan 2 1 0 1 4 4 1 5 Henrik Gustafsson 3 3 3 3 2 5 14 6 Darren Spicer 0 0 0 N 3 0 7 Alun Rossiter 3 3 1 1 3 5 11 8 Away Team No. Rider Comments 1 2 3 4 5 6 7 Rides Pts BP FM PM 1 Chris Louis 2 2 3 2 1 5 10 2 Jacek Rempala 000N 30 3 Tony Rickardsson N 3 2 2 3 3 5 13 1 4 Shane Parker 0 1 3 0 4 4 5 Zdenek Tesar 1 3 1 R 4 5 6 Ben Howe 1 1 0 0 N 4 2 1 7 David Norris 2 3 2 1 2 5 10 8 Season : 1992 Ipswich Witches 1992 - Incomplete Venue : Foxhall Stadium Competition : Challenge Ipswich 46 Indianerna 43 Date : 26/03/1992 Comments : Home Team No. -

GENERAL CATALOGUE 2019 Our History Is Our Future
Ride Regina. Be one of us! P.C13D19EC GENERAL CATALOGUE 2019 Our history is our future. The Wall of Champions. In 2019, Regina will celebrate its Innocenti’s “Lambretta”. The original Bayliss, Biaggi, Lorenzo, Rossi, by combining innovation with an 100th anniversary. The company design of Lambretta did not have a Everts, Cairoli, Herlings, Eriksson, outstanding and consistently high quality 1949 Bruno Ruffo 250cc Class 1974 Phil Read 500cc Class 1987 Jorge Martinez 80cc Class 1995 Petteri Silvan Enduro 125cc 2003 Anders Eriksson Enduro 400cc 2011 Antonio Cairoli MX1 was founded in 1919, initially transmission by chain, but after the Prado, level has allowed Regina 1950 Umberto Masetti 500cc Class 1974 M. Rathmell Trial 1987 Virginio Ferrari TT F1 1995 Anders Eriksson Enduro 350cc 2003 Juha Salminen Enduro 500cc 2011 Ken Roczen MX2 1950 Bruno Ruffo 125cc Class 1975 Paolo Pileri 125cc Class 1987 John Van Den Berk 125cc MX 1995 Jordi Tarrés Trial 2004 James Toseland WSBK 2011 Greg Hancock Speedway manufacturing chains and freewheels problems experienced with the cardan and Remes to be a market leader, 1951 Carlo Ubbiali 125cc Class 1975 Walter Villa 250cc Class 1987 Jordi Tarrés Trial 1995 Kari Tiainen Enduro 500cc 2004 Stefan Everts MX1 2012 Marc Marquez Moto 2 for bicycles. In those days, the transmission, the company asked for appreciated the enabling it to serve 1951 Bruno Ruffo 250cc Class 1975 R. Steinhausen/J. Huber Sidecar 1988 Jorge Martinez 80cc Class 1996 Max Biaggi 250cc Class 2004 Juha Salminen Enduro 2 2012 Max Biaggi WSBK -

Back to the Belle Vue!
THE FIRST EVER FREE SPEEDWAY MAGAZINE! THE VERY BEST OF ISSUE 2 – AUTUMN 2005 BACK TO THE BELLE VUE! PHOTOGRAPHS OF THE BEST SPEEDWAY TRACK IN THE WORLD…EVER! A HOME FROM HOME REG FEARMAN SHELBOURNE ARENA PIX THE POLISH PERSPECTIVE HERE TO HELP WWW.SPEEDWAYPLUS.CO.UK =EDITORIAL Welcome to the second edition of CONTENTS ‘The very best of SpeedwayPlus’. Our first issue proved to be a INTERVIEW: REG FEARMAN 3 tremendous success with several thousand copies being downloaded. Hopefully there are COLUMNIST: MIKE BENNETT 7 now a fair number of paper copies sitting on shelves just waiting to HISTORY: NO PLACE LIKE HOME 10 be rediscovered in the years ahead. COLUMNIST: DAVE GREEN 13 We’re delighted to be joined this time around by legendary motormouth Mike l TRACK PICTURES: ARENA ESSEX 14 Bennett. Mike returned to the sport in 2004 after we featured an interview IRELAND: PROMOTING IN SHELBOURNE 15 with him on the website. When the interview was published he was invited along as a guest to King’s Lynn and COLUMNIST: CHRIS SEAWARD 16 ended up presenting that night’s meeting. So I’m afraid it’s, at least TRACK PICTURES: HYDE ROAD 17 partly, our fault that he’s back! BOOK EXTRACT: CHRIS MORTON 18 Reg Fearman and Chris Morton MBE are other ‘names’ that we must thank for their contributions. Reg was kind TALL TALES: BLACKPOOL & ROME 20 enough to consent to an interview with Steve Harland and he talked freely about his time as rider and promoter. Chris has allowed us to publish a All correspondence to: lengthy extract from his new autobiography. -
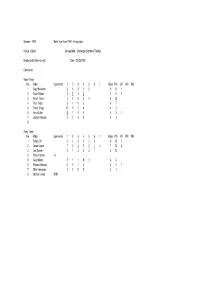
Season : 1992 Belle Vue Aces 1992 - Incomplete
Season : 1992 Belle Vue Aces 1992 - Incomplete Venue : Odsal Competition : Challenge (Northern Trophy) Bradford 45 Belle Vue 45 Date : 21/03/1992 Comments : Home Team No. Rider Comments 1 2 3 4 5 6 7 Rides Pts BP FM PM 1 Gary Havelock 2 3 3 1 0 5 9 1 2 Sean Wilson 3 2 1 2 4 8 2 3 Kelvin Tatum 3 2 3 3 2 5 13 4Paul Thorp 3103 47 5Simon Wigg R020 42 6 Kevin Little 2 1 0 0 4 3 1 7 Darren Pearson 3 0 0 0 4 3 8 Away Team No. Rider Comments 1 2 3 4 5 6 7 Rides Pts BP FM PM 1 Bobby Ott 0 1 3 3 1 2 6 10 2 2 Jason Lyons 1 2 2 2 2 1 3 7 13 4 3 Joe Screen 2 1 3 3 3 1 6 13 4 Shawn Moran r/r 5 Kelly Moran 2 1 1 N 2 4 6 6 Richard Musson 0 0 1 1 4 2 1 7 Mike Hampson 1 0 N 0 3 1 8 Michael Howe DNR Season : 1992 Belle Vue Aces 1992 - Incomplete Venue : Kirkmanshulme Lane Competition : Challenge (Northern Trophy) Belle Vue 47 Bradford 43 Date : 27/03/1992 Comments : Home Team No. Rider Comments 1 2 3 4 5 6 7 Rides Pts BP FM PM 1 Bobby Ott 1 3 3 3 1 5 11 1 2 Jason Lyons 2 2 2 2 4 8 2 3 Joe Screen 2 3 2 1 3 5 11 1 4 Frede Schott 1 0 R R 4 1 5 Kelly Moran 3 3 3 2 4 11 6 Richard Musson 3 0 0 1 1 5 5 7 Mike Hampson R 0 0 N 3 0 8 Away Team No. -

Årsracets Program
U T S T Ä L L N I N G S V I E S T A D - L I N K Ö P I N G - S W E D E N 3 1 J U L I - 2 A U G U S T I 2 0 1 5 1 ARRANGÖRER: LINKÖPINGS MS MCHK-R MCHK LAYOUT: ZiD OFFICIELLT JUBILEUMSPROGRAM 80 SEK INNEHÅLL MOTORCYKELVÄNNER Bäst i världen!? Inledning/Funktionärer ........................................................................................3 Statistik 50-Års Racet...........................................................................................4 LMS och MCHK-Racing presenterar stolt årets jubileums och ”extra-allt-utgåva” av Årsracet! Flaggsignaler .........................................................................................................7 I år firar MCHK 50-årsjubileum och därmed är det 29:e gången vi genomför Årsracet på Linköpings Motor- stadion. Flera av förare och funktionärer har varit med under hela resan från start och fått uppleva hur Tidsschema ............................................................................................................8 arrangemanget växt och utvecklats till att bli en av dom bästa classic-tävlingarna i Europa. RR Varvtider/Resultat i mobilen ..........................................................................9 Även Linköpings Motorstadion har växt och utvecklats ihop med Årsracet. I år kan vi erbjuda ett helt nytt Klassindelning RR .............................................................................................. 10 hus med speakertorn, race-office och servering med modernt kök i samma byggnad. Allt detta tack vare ER RR Klass 2B, 50cc............................................................................................. -

The French Huguenot Connection
Swedish American Genealogist Volume 36 Number 4 Article 3 12-1-2016 The French Huguenot Connection Russel Chalberg Follow this and additional works at: https://digitalcommons.augustana.edu/swensonsag Part of the Genealogy Commons, and the Scandinavian Studies Commons Recommended Citation Chalberg, Russel (2016) "The French Huguenot Connection," Swedish American Genealogist: Vol. 36 : No. 4 , Article 3. Available at: https://digitalcommons.augustana.edu/swensonsag/vol36/iss4/3 This Article is brought to you for free and open access by the Swenson Swedish Immigration Research Center at Augustana Digital Commons. It has been accepted for inclusion in Swedish American Genealogist by an authorized editor of Augustana Digital Commons. For more information, please contact [email protected]. The French Huguenot Connection A Swenson Family Legend BY RUSSEL CHALBERG <[email protected]> For at least the last two generations, the In a tape recording, made many years Swenson family has circulated an oral story ago, Ruth Swenson (b. 2 Mar. 1885) states that this family was not 100% Swedish; it very clearly, “that one of her ancestors was is said to have a bit of French blood. A re- a Huguenot who had escaped the per- cent DNA test seems to confirm this fact secution in France.” as the test indicated approximately 2% to Ellen Ruth Swenson was the youngest be Western European. of nine children born to Frans August Sven- As this family legend goes, one of their son (b. 10 Mar. 1838) and Johanna Johans- ancestors was a Huguenot who came to dotter (b. 9 Feb. 1840). This family lived Sweden in order to escape religious per- in Ljuder parish (Kron.), until the father, secution in France. -

Season : 1992 Wolverhampton Wolves 1992 - Incomplete
Season : 1992 Wolverhampton Wolves 1992 - Incomplete Venue : Monmore Green Competition : Challenge Wolverhampton 51 Indianerna 38 Date : 23/03/1992 Comments : Home Team No. Rider Comments 1 2 3 4 5 6 7 Rides Pts BP FM PM 1 Ronnie Correy 2 2 3 3 4 10 2 Charles Ermolenko 1 1 2 2 4 6 3 3 Sam Ermolenko 2 3 3 3 2 5 13 4 Graham Jones 3 3 1 2 0 5 9 5 Peter Karlsson FX1 fn N 2 1 6 Wayne Carter 3 1 2 2 1 5 9 3 7 Stephen Morris 1 1 0 FX 1 N 5 3 1 8 Away Team No. Rider Comments 1 2 3 4 5 6 7 Rides Pts BP FM PM 1 Henrik Gustafsson 3 2 3 2 2 1 6 13 2 Stefan Danno 0 0 0 1 4 1 1 3 Bo Petersen 3 3 3 2 3 3 6 17 4 Magnus Oscarsson F 0 1 0 4 1 5 Dennis Lofqvist fn N N N 0 0 6 Tommy Lindgren 2 2 0 1 0 5 5 7 Mikael Haglund 0 0 1 N 0 0 5 1 8 Season : 1992 Wolverhampton Wolves 1992 - Incomplete Venue : Monmore Green Competition : Gold Cup Wolverhampton 47 Cradley Heath 43 Date : 06/04/1992 Comments : Home Team No. Rider Comments 1 2 3 4 5 6 7 Rides Pts BP FM PM 1 Ronnie Correy 2 2 3 2 0 5 9 2 Charles Ermolenko 1 1 2 2 4 6 3 3 Sam Ermolenko 2 3 3 3 3 5 14 4 Graham Jones 2 2 1 X 4 5 1 5 Peter Karlsson 1 1 1 0 4 3 2 6 Wayne Carter 2 1 2 0 2 5 7 3 7 Stephen Morris 3 FX 0 N 3 3 8 Away Team No. -

Tiskova Informace
PARDUBICKÝ PLOCHODRÁŽNÍ VÍKEND 27. - 29. 9. 2013 Pardubické plochodrážní závody mají ve sv ětě obrovský zvuk. Jsou založeny nejenom na dlouholeté tradici, ale p ředevším na kvalit ě. Proto lákají pravideln ě na start jezdce nejenom zvu čných jmen, ale i ty, jímž kariéra teprve za číná. To p řitahuje každoro čně spousty p říznivc ů a to vše dohromady d ělá z Pardubic o posledním zá řijovém víkendu složeném s těchto čty ř závod ů hlavní m ěsto sv ětové ploché dráhy. Čty ři tak velké plochodrážní akce lze vid ět z jediný víkend na jediném míst ě na sv ětě - ve Svítkov ě ZLATÁ P ŘILBA V Pardubicích se od roku 1929 koná motocyklový závod na ploché dráze. V sou časnosti je nejstarším pravideln ě po řádaným závodem tohoto druhu na sv ětě. Každoro čně láká do hledišt ě stadionu ve Svítkov ě více jak deset tisíc p říznivc ů, kterým nabízí atraktivní podívanou na šestici jezdc ů jedoucích bezmála stokilometrovou rychlostí po ovále na strojích, které nemají brzdy. Na hlinitopís čitém povrchu se p ředstaví op ět mnoho jezdc ů absolutní sv ětové špi čky bojujících o ve sv ětě cen ěnou trofej. Proti nim stojí domácí jezdci, ti čekající na úsp ěch už dlouhých šestnáct let. Tak t ěžké je se zde prosadit, tak kvalitní jezdí do Pardubic konkurence. FINÁLE MISTROVSTVÍ SV ĚTA DRUŽSTEV NA PLOCHÉ DRÁZE DO 21 LET V p ředve čer slavného plochodrážního závodu o Zlatou p řilbu se v Pardubicích uskute ční finálový závod ů mistrovství sv ěta družstev v kategorii jezdc ů do 21 let. -

Tritius/WORK/6909/Jan Moroviec.Pdf
Vyšší odborná škola publicistiky AVD média Jan Mroviec 20 let čekání na českého vítěze Zlaté přilby města Pardubic Absolventská práce Praha 2017 Vedoucí práce: Mgr. Roman Bradáč Prohlášení Prohlašuji, že jsem absolventskou práci „20 let na čekání českého vítěze Zlaté přilby města Pardubic“ vypracoval samostatně. Veškerou použitou literaturu a další podkladové materiály uvádím v přiloženém seznamu literatury. V Praze, dne ......................................... , Podpis autora: ..................................................................... - 2 - Poděkování V první řadě bych chtěl poděkovat za vedení absolventské práce Romanu Bradáčovi, který se ke mně neotočil zády ani přes svoji výraznou časovou vytíženost. Velký dík patří předsedovi plochodrážního svazu české republiky Petru Moravcovi, který mi umožnil přístup do klubového archívu AMK Zlatá přilba Pardubice a zapůjčením vlastních dokumentů mě správně navedl. Nejen Petru Moravcovi, ale i Evženu Erbanovi, Jiřímu Štanclovi a Václavu Milíkovi bych rád poděkoval za jejich odborné názory, bez kterých by tato práce nemohla vzniknout. Poděkování míří i k sportovnímu novináři Martinu Kézrovi, který mi poskytl zpětnou vazbu k mé práci. - 3 - Abstrakt Tématem absolventské práce je doba, během níž nevyhrál nejstarší plochodrážní závod – Zlatou přilbu města Pardubic – žádný český závodník. Práce si klade za cíl popsat hlavní příčiny českých neúspěchů, shrnout, jaké jsou v současnosti naděje českých jezdců na vítězství v závodu a zjistit, zda se pořadatelé obávají zániku prestižní akce za předpokladu, že čeští jezdci nebudou trofej vyhrávat. V praktické i teoretické části pomocí výpovědí plochodrážních expertů a odborné literatury. Výsledek práce ukázal, že hlavní příčina českých neúspěchů v závodu o Zlatou přilbu města Pardubic je hlavně ve změně podmínek po revoluci, s kterými se domácí jezdci nedokázali dobře vypořádat. Odborníci v současnosti poukazují na jediného českého závodníka, který by měl reálnou šanci závod vyhrát a pořadatelé se ztráty zájmu ze strany diváků nebojí.