Enfortumab Vedotin-Ejfv) for Injection, for Intravenous Use Monitor Patients for Signs Or Symptoms of Ocular Disorders
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Antibody–Drug Conjugates
Published OnlineFirst April 12, 2019; DOI: 10.1158/1078-0432.CCR-19-0272 Review Clinical Cancer Research Antibody–Drug Conjugates: Future Directions in Clinical and Translational Strategies to Improve the Therapeutic Index Steven Coats1, Marna Williams1, Benjamin Kebble1, Rakesh Dixit1, Leo Tseng1, Nai-Shun Yao1, David A. Tice1, and Jean-Charles Soria1,2 Abstract Since the first approval of gemtuzumab ozogamicin nism of activity of the cytotoxic warhead. However, the (Mylotarg; Pfizer; CD33 targeted), two additional antibody– enthusiasm to develop ADCs has not been dampened; drug conjugates (ADC), brentuximab vedotin (Adcetris; Seat- approximately 80 ADCs are in clinical development in tle Genetics, Inc.; CD30 targeted) and inotuzumab ozogami- nearly 600 clinical trials, and 2 to 3 novel ADCs are likely cin (Besponsa; Pfizer; CD22 targeted), have been approved for to be approved within the next few years. While the hematologic cancers and 1 ADC, trastuzumab emtansine promise of a more targeted chemotherapy with less tox- (Kadcyla; Genentech; HER2 targeted), has been approved to icity has not yet been realized with ADCs, improvements treat breast cancer. Despite a clear clinical benefit being dem- in technology combined with a wealth of clinical data are onstrated for all 4 approved ADCs, the toxicity profiles are helping to shape the future development of ADCs. In this comparable with those of standard-of-care chemotherapeu- review, we discuss the clinical and translational strategies tics, with dose-limiting toxicities associated with the mecha- associated with improving the therapeutic index for ADCs. Introduction in antibody, linker, and warhead technologies in significant depth (2, 3, 8, 9). Antibody–drug conjugates (ADC) were initially designed to leverage the exquisite specificity of antibodies to deliver targeted potent chemotherapeutic agents with the intention of improving Overview of ADCs in Clinical Development the therapeutic index (the ratio between the toxic dose and the Four ADCs have been approved over the last 20 years (Fig. -

Genmab and Seattle Genetics Present Data from Tisotumab Vedotin Innovatv 204 Pivotal Trial in Recurrent Or Metastatic Cervical Cancer at ESMO Virtual Congress 2020
Genmab and Seattle Genetics Present Data from Tisotumab Vedotin innovaTV 204 Pivotal Trial in Recurrent or Metastatic Cervical Cancer at ESMO Virtual Congress 2020 Media Release COPENHAGEN, Denmark and BOTHELL, Wash., 21 September 2020 • Data featured in late-breaking proffered paper oral presentation • Biologics license application submission planned to support accelerated approval pathway with the FDA Genmab A/S (Nasdaq: GMAB) and Seattle Genetics, Inc. (Nasdaq: SGEN) today presented data from the innovaTV 204 pivotal phase 2, single-arm clinical trial evaluating tisotumab vedotin as monotherapy in patients with previously treated recurrent and/or metastatic cervical cancer at the European Society for Medical Oncology (ESMO) Virtual Congress 2020. Patients had previously received a doublet chemotherapy and, if eligible, bevacizumab as first-line therapy. Results from the trial showed a 24 percent confirmed objective response rate (ORR) by independent central review with a median duration of response (DOR) of 8.3 months. The most common treatment-related adverse events (greater than or equal to 20 percent) included alopecia, epistaxis (nose bleeds), nausea, conjunctivitis, fatigue and dry eye. Tisotumab vedotin is an investigational antibody-drug conjugate (ADC) directed to tissue factor (TF), which is prevalent on solid tumors including cervical cancer and can promote tumor growth, angiogenesis and metastasis.1 Current therapies for previously treated recurrent and/or metastatic cervical cancer generally result in limited objective response rates of typically less than 15 percent with median overall survival ranging from 6.0 to 9.4 months.1-8 “Following resistance to or progression on first-line standard of care therapy, there are limited treatment options for women with metastatic cervical cancer,” said Robert L. -
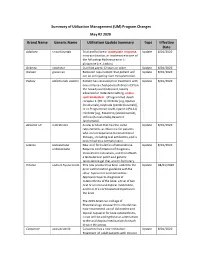
Summary of Utilization Management (UM) Program Changes May #2 2020
Summary of Utilization Management (UM) Program Changes May #2 2020 Brand Name Generic Name Utilization Update Summary Type Effective Date Adakveo crizanlizumab Trial and failure or inadequate response, Update 8/01/2020 contraindication, or intolerance to one of the following: Hydroxyurea or L- glutamine (i.e., Endari) Oxbryta voxelotor Clarified age to 12 years or older Update 8/01/2020 Givlaari givosiran Removed requirement that patient will Update 8/01/2020 not be anticipating liver transplantation. Padcev enfortumab vedotin Patient has received prior treatment with Update 8/01/2020 one immune checkpoint inhibitors (CPI) in the neoadjuvant/adjuvant, locally advanced or metastatic setting, unless contraindicated: i) Programmed death receptor-1 (PD-1) inhibitor [e.g.,Opdivo (nivolumab), Keytruda (pembrolizumab)], or ii) Programmed death-ligand 1 (PD-L1) inhibitor [e.g., Tecentriq (atezolizumab), Imfinzi (durvalumab), Bavencio (avelumab)]. Absorica LD isotretinoin A new product that has the same Update 8/01/2020 requirements as Absorica: for patients who are unresponsive to conventional therapy, including oral antibiotics, and is prescribed by a dermatologist. Jatenzo testosterone New oral formulation of testosterone. Update 8/01/2020 undecanoate Requires confirmation of diagnosis, testosterone lab values, and trial of both a testosterone patch and generic testosterone gel that are on formulary. Triluron sodium hyaluronate This new product has been added to the Update 08/01/2020 prior authorization guideline with the other hyaluronic acid derivatives. Approval requires diagnosis of osteoarthritis of the knee, a trial of two oral or an oral and topical medication, and trial of a corticosteroid injection in the knee. The 2019 American College of Rheumatology Osteoarthritis Guidelines now recommend use of duloxetine and topical capsaicin for knee osteoarthritis, so we will be adding these as alternatives to the oral/topical medications for each drug in this group. -

Antibodies for the Treatment of Brain Metastases, a Dream Or a Reality?
pharmaceutics Review Antibodies for the Treatment of Brain Metastases, a Dream or a Reality? Marco Cavaco, Diana Gaspar, Miguel ARB Castanho * and Vera Neves * Instituto de Medicina Molecular, Faculdade de Medicina, Universidade de Lisboa, Av. Prof. Egas Moniz, 1649-028 Lisboa, Portugal * Correspondence: [email protected] (M.A.R.B.C.); [email protected] (V.N.) Received: 19 November 2019; Accepted: 28 December 2019; Published: 13 January 2020 Abstract: The incidence of brain metastases (BM) in cancer patients is increasing. After diagnosis, overall survival (OS) is poor, elicited by the lack of an effective treatment. Monoclonal antibody (mAb)-based therapy has achieved remarkable success in treating both hematologic and non-central-nervous system (CNS) tumors due to their inherent targeting specificity. However, the use of mAbs in the treatment of CNS tumors is restricted by the blood–brain barrier (BBB) that hinders the delivery of either small-molecules drugs (sMDs) or therapeutic proteins (TPs). To overcome this limitation, active research is focused on the development of strategies to deliver TPs and increase their concentration in the brain. Yet, their molecular weight and hydrophilic nature turn this task into a challenge. The use of BBB peptide shuttles is an elegant strategy. They explore either receptor-mediated transcytosis (RMT) or adsorptive-mediated transcytosis (AMT) to cross the BBB. The latter is preferable since it avoids enzymatic degradation, receptor saturation, and competition with natural receptor substrates, which reduces adverse events. Therefore, the combination of mAbs properties (e.g., selectivity and long half-life) with BBB peptide shuttles (e.g., BBB translocation and delivery into the brain) turns the therapeutic conjugate in a valid approach to safely overcome the BBB and efficiently eliminate metastatic brain cells. -

Enfortumab Vedotin Antibody-Drug Conjugate Targeting Nectin-4 Is a Highly Potent
Author Manuscript Published OnlineFirst on March 24, 2016; DOI: 10.1158/0008-5472.CAN-15-1313 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Enfortumab Vedotin Antibody-Drug Conjugate Targeting Nectin-4 is a Highly Potent Therapeutic Agent in Multiple Preclinical Cancer Models Pia M. Challita-Eid1*, Daulet Satpayev2, Peng Yang1, Zili An1, Karen Morrison1, Yuriy Shostak1, Arthur Raitano1, Rossana Nadell1, Wendy Liu1, Dawn Ratay Lortie1, Linnette Capo1, Alla Verlinsky1, Monica Leavitt1, Faisal Malik1, Hector Aviña1, Claudia I. Guevara1, Nick Dinh1, Sher Karki1, Banmeet S. Anand1, Daniel S. Pereira1, Ingrid B. J. Joseph1, Fernando Doñate1, Kendall Morrison1, David R. Stover1 1Agensys Inc., Santa Monica, CA 2Previously at Agensys, Inc.; currently at ImaginAb, Inglewood, CA All authors worked at Agensys Inc. during the period of the study. *Corresponding author: Pia M. Challita-Eid, Agensys, Inc., 1800 Stewart Street, Santa Monica, CA 90404; Phone: (424) 280-5000; Email: [email protected] Financial support, including the source and number of grants, for each author: Funded by Agensys, Inc., an affiliate of Astellas Pharma, Inc. Disclosure of Potential Conflicts of Interest: All authors are/were employees of Agensys, Inc., an affiliate of Astellas Pharma, Inc., during the period of the study. Running title: ADC cancer therapeutic targeting nectin-4 Keywords: antibody, xenograft, nectin, cancer, ADC Text word count: 4564 Abstract word count: 227 Total number of figures/tables: 6 figures and 1 table 1 Downloaded from cancerres.aacrjournals.org on September 23, 2021. © 2016 American Association for Cancer Research. Author Manuscript Published OnlineFirst on March 24, 2016; DOI: 10.1158/0008-5472.CAN-15-1313 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. -

Genmab and Seagen Submit Tisotumab Vedotin Biologics License Application to the U.S
Genmab and Seagen Submit Tisotumab Vedotin Biologics License Application to the U.S. FDA for Patients with Recurrent or Metastatic Cervical Cancer Company Announcement • Submission based on positive pivotal innovaTV 204 trial results presented at the European Society of Medical Oncology Virtual Congress 2020 COPENHAGEN, Denmark and BOTHELL, Wash.; Feb 10, 2021 – Genmab A/S (Nasdaq: GMAB) and Seagen Inc. (Nasdaq: SGEN) today announced the submission of a Biologics License Application (BLA) to the U.S. Food and Drug Administration (FDA) seeking accelerated approval for tisotumab vedotin. This BLA requests FDA approval of tisotumab vedotin for the treatment of patients with recurrent or metastatic cervical cancer with disease progression on or after chemotherapy. The submission is based on the results of the innovaTV 204 pivotal phase 2 single-arm clinical trial evaluating tisotumab vedotin as monotherapy in this setting. The topline results from the phase 2 study were announced in June 2020 and data were presented at the European Society for Medical Oncology (ESMO) Virtual Congress 2020. Tisotumab vedotin is an investigational antibody-drug conjugate (ADC) directed to tissue factor (TF), a cell-surface protein expressed on multiple solid tumors including cervical cancer and is associated with tumor growth, angiogenesis, metastasis and poor prognosis.1 “This BLA submission is an important step toward our goal of improving the lives of women with recurrent or metastatic cervical cancer. I would like to thank the patients, nurses, physicians and researchers who participated in the innovaTV 204 trial, which is the basis of this submission,” said Jan van de Winkel, Ph.D., Chief Executive Officer of Genmab. -

Antibody–Drug Conjugates for Cancer Therapy
molecules Review Antibody–Drug Conjugates for Cancer Therapy Umbreen Hafeez 1,2,3, Sagun Parakh 1,2,3 , Hui K. Gan 1,2,3,4 and Andrew M. Scott 1,3,4,5,* 1 Tumour Targeting Laboratory, Olivia Newton-John Cancer Research Institute, Melbourne, VIC 3084, Australia; [email protected] (U.H.); [email protected] (S.P.); [email protected] (H.K.G.) 2 Department of Medical Oncology, Olivia Newton-John Cancer and Wellness Centre, Austin Health, Melbourne, VIC 3084, Australia 3 School of Cancer Medicine, La Trobe University, Melbourne, VIC 3084, Australia 4 Department of Medicine, University of Melbourne, Melbourne, VIC 3084, Australia 5 Department of Molecular Imaging and Therapy, Austin Health, Melbourne, VIC 3084, Australia * Correspondence: [email protected]; Tel.: +61-39496-5000 Academic Editor: João Paulo C. Tomé Received: 14 August 2020; Accepted: 13 October 2020; Published: 16 October 2020 Abstract: Antibody–drug conjugates (ADCs) are novel drugs that exploit the specificity of a monoclonal antibody (mAb) to reach target antigens expressed on cancer cells for the delivery of a potent cytotoxic payload. ADCs provide a unique opportunity to deliver drugs to tumor cells while minimizing toxicity to normal tissue, achieving wider therapeutic windows and enhanced pharmacokinetic/pharmacodynamic properties. To date, nine ADCs have been approved by the FDA and more than 80 ADCs are under clinical development worldwide. In this paper, we provide an overview of the biology and chemistry of each component of ADC design. We briefly discuss the clinical experience with approved ADCs and the various pathways involved in ADC resistance. -

Oncology Dose Reduction Program Beginning July 1, 2021
Medicare Advantage Provider Bulletin February 2021 Oncology Dose Reduction Program beginning July 1, 2021 Simply Healthcare Plans, Inc. (Simply) is committed to being a valued healthcare partner in identifying ways to achieve better health outcomes, lower costs and deliver access to better healthcare experiences for consumers. Effective for dates of service on or after July 1, 2021, providers for our Medicare Advantage plan members covered by Simply will be asked in selective circumstances to voluntarily reduce the requested dose to the nearest whole vial for over 40 oncology medications, listed below. Reviews for these oncology drugs will continue to be administered by the reviewing company, either AIM Specialty Health®* or IngenioRx.* Providers will be asked whether or not they will accept the dose reduction at the initial review point in the prior authorization process. Within the provider portal, a pop-up question will appear related to dose reduction. If the patient is considered unable to have his or her dose reduced, then a second question will appear asking for the provider’s clinical reasoning. For requests made outside of the provider portal (for example, called-in or faxed-in prior authorization requests), the same questions will be asked by the registered nurse or medical director who is reviewing the request. Since this program is voluntary, the decision made regarding dose reduction will not affect the final decision on the prior authorization. The dose reduction questions will appear only if the originally requested dose is within 10% of the nearest whole vial. This threshold is based on current medical literature and recommendations from the Hematology and Oncology Pharmacists Association (HOPA) that it is appropriate to consider dose rounding within 10%. -

Enfortumab Vedotin for Locally Advanced Or Metastatic Urothelial
Author Manuscript Published OnlineFirst on September 22, 2020; DOI: 10.1158/1078-0432.CCR-20-2275 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Title: FDA Approval Summary: Enfortumab Vedotin for Locally Advanced or Metastatic Urothelial Carcinoma Authors: Elaine Chang1, Chana Weinstock1, Lijun Zhang1, Rosane Charlab1, Sarah E. Dorff1, Yutao Gong1, Vicky Hsu1, Fang Li1, Tiffany K. Ricks1, Pengfei Song1, Shenghui Tang1, Peter E. Waldron1, Jingyu Yu1, Eias Zahalka1, Kirsten B. Goldberg2, Richard Pazdur1,2, Marc R. Theoret1,2, Amna Ibrahim1, Julia A. Beaver1 Authors’ Affiliations: 1Center for Drug Evaluation and Research, U.S. Food and Drug Administration; 2Oncology Center of Excellence, U.S. Food and Drug Administration Running Title: FDA Approval Summary: Enfortumab vedotin Corresponding Author: Elaine Chang, Office of Oncologic Diseases, CDER, U.S. Food and Drug Administration, WO22 Room 2169, 10903 New Hampshire Avenue, Silver Spring, MD 20993. Phone 240-402-2628. Fax: 301-796-9909. Email: [email protected]. Note: This is a U.S. Government work. There are no restrictions on its use. Disclosure of Potential Conflicts of Interest: The authors report no financial interests or relationships with the commercial sponsors of any products discussed in this report. Word count: Abstract 200; Text 2996; Tables = 3; Figures = 1 ; References = 18 1 Downloaded from clincancerres.aacrjournals.org on September 26, 2021. © 2020 American Association for Cancer Research. Author Manuscript Published OnlineFirst on September 22, 2020; DOI: 10.1158/1078-0432.CCR-20-2275 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. -

Will These 12 Cancer Trials Change Clinical Practice? Asco 2019 and Future Directions in Oncology | 2
WILL THESE 12 CANCER TRIALS CHANGE CLINICAL PRACTICE? ASCO 2019 AND FUTURE DIRECTIONS IN ONCOLOGY | 2 Contents Introduction 3 Bladder cancer – Enfortumab vedotin from Seattle Genetics and Astellas 4 Breast cancer – Margetuximab from MacroGenics and Kisqali from Novartis 5 Gastric cancer – Keytruda from Merck & Co 7 Lymphoma – Yescarta from Kite Pharma 8 Lung cancer – BLU-667 from Blueprint Medicines 9 Myeloma – Darzalex from Janssen and isatuximab from Sanofi 9 Pancreatic cancer – Lynparza from AstraZeneca and Merck & Co 10 Prostate cancer – Xtandi from Pfizer and Astellas and Erleada from Johnson & Johnson 11 Multiple indications – AMG-510 from Amgen 12 Conclusion 14 References 15 © 2019 Kantar www.kantar.com/health | 3 Introduction The American Society of Clinical Oncology’s annual meeting in Chicago is the preeminent date on the cancer research calendar, and ASCO 2019 was no exception. The healthcare professionals, pharmaceutical industry executives and global media that assembled at McCormick Place in June were presented with a wealth of new clinical data readouts from the oncology spectrum. Kantar was on-site in Chicago gathering insights on the future direction for oncology. As the dust settles on ASCO 2019, and clinical practice begins to adapt to this year’s meeting, this report picks out the key findings from 12 cancer trials, and we put the ASCO headlines into context for patients, prescribers and the pharmaceutical industry. In doing so, our experts examine trial highlights from some of the biggest players in the business as well as those seeking to make a name for themselves. Looking at presentations by AstraZeneca, Gilead, Janssen, Merck & Co, Novartis, Seattle Genetics and others, we also dig into the robustness of the data and, for some medicines, find compelling levels of benefit. -

Immunotherapy Combinations and Sequences in Urothelial Cancer: Facts and Hopes Alejo Rodriguez-Vida1, Jose Luis Perez-Gracia2, and Joaquim Bellmunt1,3
Published OnlineFirst July 10, 2018; DOI: 10.1158/1078-0432.CCR-17-3108 Review Clinical Cancer Research Immunotherapy Combinations and Sequences in Urothelial Cancer: Facts and Hopes Alejo Rodriguez-Vida1, Jose Luis Perez-Gracia2, and Joaquim Bellmunt1,3 Abstract Immune checkpoint inhibitors (ICI) have emerged as a maintained remissions. Active research is ongoing in sev- novel therapeutic strategy that achieves significant clinical eral fields, aiming to increase the number of patients that benefit in several tumor types, including urothelial cancer. benefit from ICI, and this research is largely based on the Overall, these agents have shown objective response rates development of biomarkers for personalized immunother- of around 20% to 23%, which indicates that a significant apy and novel combinations of ICI with other agents. proportion of patients do not benefit from immunother- This article will review ongoing efforts to develop combi- apy when given as monotherapy. Moreover, despite an nations of ICI with other therapeutic strategies in patients initial response to therapy and an improvement in the with urothelial cancer, including chemotherapy, targeted median duration of response compared with chemother- agents, other immunotherapy strategies, and radiotherapy. apy, still only half of the patients develop long-term Clin Cancer Res; 24(24); 6115–24. Ó2018 AACR. Introduction urothelial cancer who are ineligible for cisplatin (8, 9), thus leading to ongoing randomized phase III trials in this setting. Immune checkpoint inhibitors (ICI) have become a major step Overall, these studies have shown objective response rates forward in the treatment of urothelial cancer. Indeed, significant (ORR) of around 20%–23%, which indicates that a significant clinical activity has been demonstrated since the first studies were proportion of patient does not benefit from immunotherapy reported in this tumor type, which also highlighted for the first when given as monotherapy. -

Antibody Drug Conjugates: Future Directions in Clinical and Translational Strategies to 2 Improve the Therapeutic Index
Author Manuscript Published OnlineFirst on April 12, 2019; DOI: 10.1158/1078-0432.CCR-19-0272 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. 1 1 Antibody Drug Conjugates: Future Directions in Clinical and Translational Strategies to 2 Improve the Therapeutic Index 3 4 Authors: Steven Coats1, Marna Williams1, Benjamin Kebble1, Rakesh Dixit1, Leo Tseng1, Nai- 5 Shun Yao1, David A. Tice1, and Jean-Charles Soria1,2 6 7 Affiliations: 1AstraZeneca, Gaithersburg, MD, USA. 2University Paris-Sud, Orsay, France. 8 9 Running title: Advances in Antibody Drug Conjugate Clinical Development 10 11 Corresponding Author: Steven Coats 12 Research and Development Oncology 13 AstraZeneca 14 1 MedImmune Way 15 Gaithersburg, MD 20878 16 Email: [email protected] 17 18 DISCLOSURE OF POTENTIAL CONFLICT OF INTEREST 19 All authors are employees of AstraZeneca and hold stock/stock options in AstraZeneca. Dr. 20 Soria also holds stock/stock options in Gritstone. Over the last 5 years, Dr. Soria has received 21 consultancy fees from AstraZeneca, Astex, Clovis, GSK, GamaMabs, Lilly, MSD, Mission 22 Therapeutics, Merus, Pfizer, PharmaMar, Pierre Fabre, Roche/Genentech, Sanofi, Servier, 23 Symphogen, and Takeda. 24 25 26 Downloaded from clincancerres.aacrjournals.org on September 28, 2021. © 2019 American Association for Cancer Research. Author Manuscript Published OnlineFirst on April 12, 2019; DOI: 10.1158/1078-0432.CCR-19-0272 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. 2 27 ABSTRACT 28 Since the first approval of gemtuzumab ozogamicin (Mylotarg; CD33 targeted), 2 additional 29 antibody drug conjugates (ADCs)—brentuximab vedotin (Adcetris; CD30 targeted) and 30 inotuzumab ozogamicin (Besponsa; CD22 targeted)—have been approved for hematologic 31 cancers and 1 ADC, trastuzumab emtansine (Kadcyla; HER2 targeted), has been approved to 32 treat breast cancer.