Nonlinear Optics
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Tutorial on Nonlinear Optics
Proceedings of the International School of Physics “Enrico Fermi” Course 190 “Frontiers in Modern Optics”, edited by D. Faccio, J. Dudley and M. Clerici (IOS, Amsterdam; SIF, Bologna) 2016 DOI 10.3254/978-1-61499-647-7-31 Tutorial on nonlinear optics S. Choudhary School of Electrical Engineering and Computer Science, University of Ottawa Ottawa, Ontario, K1N 6N5 Canada R. W. Boyd School of Electrical Engineering and Computer Science, University of Ottawa Ottawa, Ontario, K1N 6N5 Canada The Institute of Optics, University of Rochester Rochester, New York, 14627 USA Department of Physics, University of Ottawa Ottawa, Ontario, K1N 6N5 Canada Summary. — Nonlinear optics deals with phenomena that occur when a very intense light interacts with a material medium, modifying its optical properties. Shortly after the demonstration of first working laser in 1960 by Maiman (Nature, 187 (1960) 493), the field of nonlinear optics began with the observation of second harmonic by Franken et al. in 1961 (Phys. Rev. Lett., 7 (1961) 118). Since then, the interest in this field has grown and various nonlinear optical effects are utilized for purposes such as nonlinear microscopy, switching, harmonic generation, parametric downconversion, filamentation, etc. We present here a brief overview of the various aspects on nonlinear optics and some of the recent advances in the field. c Societ`a Italiana di Fisica 31 32 S. Choudhary and R. W. Boyd 1. – Introduction to nonlinear optics Accordingˇ¡proofsAuthor please note that we have written in full the reference quota- tions in the abstract and reordered those in the text accordingly, following the numerical sequence. -
Nonlinear Optics (Wise 2018/19) Lecture 6: November 18, 2018 5.2 Electro-Optic Amplitude Modulator 5.3 Electro-Optic Phase Modulator 5.4 Microwave Modulator
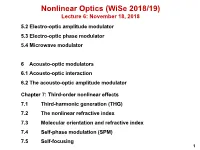
Nonlinear Optics (WiSe 2018/19) Lecture 6: November 18, 2018 5.2 Electro-optic amplitude modulator 5.3 Electro-optic phase modulator 5.4 Microwave modulator 6 Acousto-optic modulators 6.1 Acousto-optic interaction 6.2 The acousto-optic amplitude modulator Chapter 7: Third-order nonlinear effects 7.1 Third-harmonic generation (THG) 7.2 The nonlinear refractive index 7.3 Molecular orientation and refractive index 7.4 Self-phase modulation (SPM) 7.5 Self-focusing 1 5.2 Electro-optic amplitude modulator How is phase retardation converted into a amplitude modulation? 5.2. ELECTRO-OPTIC AMPLITUDE MODULATOR 93 0 direction input polarizer wave plate output polarizer Figure 5.7: Transversal electro-optic amplitude modulator from LiNbO3. Here, ∆φWP is the phase retardation due to the field-independent birefrin- 2 gence or due to an additional wave plate as shown in Fig. 5.7, and a is a co- efficient describing the relationship between field-dependent phase retardation and applied voltage. Usually we use β =45◦ to achieve 100 % transmission Iout 1 = 1 cos [∆φWP + aV (t)] (5.43) Iin 2 { − } 1 = 1 cos ∆φ cos [aV (t)] + sin ∆φ sin [aV (t)] . 2 { − WP WP } There are various applications for modulators. If the transmission through the modulator should be linearly dependent on the applied voltage, we use a bias ∆φ = π/2andobtainforaV 1 (see also Fig. 5.8) WP ≪ I 1 out = [1 + aV (t)] . (5.44) Iin 2 For a sinusoidal voltage V (t)=V0 sin ωmt (5.45) and constant input intensity, we obtain a sinusoidally varying output intensity Iout 1 = (1 + aV0 sin ωmt) . -

The Mathematics of Nonlinear Optics
The Mathematics of Nonlinear Optics Guy Metivier´ ∗ March 7, 2009 Contents 1 Introduction 4 2 Examples of equations arising in nonlinear optics 11 3 The framework of hyperbolic systems 18 3.1 Equations . 18 3.2 The dispersion relation & polarization conditions . 20 3.3 Existence and stability . 23 3.4 Continuation of solutions . 26 3.5 Global existence . 27 3.6 Local results . 29 4 Equations with parameters 32 4.1 Singular equations . 32 4.1.1 The weakly non linear case . 33 4.1.2 The case of prepared data . 34 4.1.3 Remarks on the commutation method . 38 4.1.4 Application 1 . 40 4.1.5 Application 2 . 41 4.2 Equations with rapidly varying coefficients . 45 ∗Universit´eBordeaux 1, IMB UMR 5251, 351 Cours de la Lib´eration,33405 Talence cedex, [email protected] 1 5 Geometrical Optics 49 5.1 Linear geometric optics . 49 5.1.1 An example using Fourier synthesis . 49 5.1.2 The BKW method and formal solutions . 51 5.1.3 The dispersion relation and phases . 52 5.1.4 The propagator of amplitudes . 53 5.1.5 Construction of WKB solutions . 60 5.1.6 Approximate solutions . 62 5.1.7 Exact solutions . 63 5.2 Weakly nonlinear geometric optics . 64 5.2.1 Asymptotic equations . 65 5.2.2 The structure of the profile equations I: the dispersive case . 67 5.2.3 The structure of the profile equation II : the nondispersive case; the generic Burger's eqation . 70 5.2.4 Approximate and exact solutions . -

Nonlinear Optics: the Next Decade
brought to you by View metadata, citation and similar papers at core.ac.uk CORE provided by The Australian National University Nonlinear optics: The next decade Yuri S. Kivshar Center for Ultra-high bandwidth Devices for Optical Systems (CUDOS), Nonlinear Physics Center, Research School of Physical Sciences and Engineering, Australian National University, Canberra ACT 0200, Australia Abstract: This paper concludes the Focus Serial assembled of invited papers in key areas of nonlinear optics (Editors: J.M. Dudley and R.W. Boyd), and it discusses new directions for future research in this field. © 2008 Optical Society of America OCIS codes: 190.0190 Nonlinear optics References and links 1. J.M. Dudley and R.W. Boyd, “Focus Series: Frontier of Nonlinear Optics. Introduction,” Optics Express 15, 5237-5237 (2007); http://www.opticsinfobase.org/oe/abstract.cfm?URI=oe-15-8-5237 Nonlinear optics describes the behavior of light in media with nonlinear response. Its tradi- tional topics cover different types of parametric processes, such as second-harmonic generation, as well as a variety of self-action effects, such as filamentation and solitons, typically observed at high light intensities delivered by pulsed lasers. While the study of nonlinear effects has a very long history going back to the physics of mechanical systems, the field of nonlinear op- tics is relatively young and, as a matter of fact, was born only after the invention of the laser. Soon after, the study of light-matter interaction emerged as an active direction of research and boosted the developments in material science and source technologies. Nowadays, nonlinear optics has evolved into many different branches, depending on the form of the material used for studying the nonlinear phenomena. -

Rogue-Wave Solutions for an Inhomogeneous Nonlinear System in a Geophysical fluid Or Inhomogeneous Optical Medium
Commun Nonlinear Sci Numer Simulat 36 (2016) 266–272 Contents lists available at ScienceDirect Commun Nonlinear Sci Numer Simulat journal homepage: www.elsevier.com/locate/cnsns Rogue-wave solutions for an inhomogeneous nonlinear system in a geophysical fluid or inhomogeneous optical medium Xi-Yang Xie a, Bo Tian a,∗, Yan Jiang a, Wen-Rong Sun a, Ya Sun a, Yi-Tian Gao b a State Key Laboratory of Information Photonics and Optical Communications, and School of Science, Beijing University of Posts and Telecommunications, Beijing 100876, China b Ministry-of-Education Key Laboratory of Fluid Mechanics and National Laboratory for Computational Fluid Dynamics, Beijing University of Aeronautics and Astronautics, Beijing 100191, China article info abstract Article history: Under investigation in this paper is an inhomogeneous nonlinear system, which describes Received 23 September 2015 the marginally-unstable baroclinic wave packets in a geophysical fluid or ultra-short pulses Accepted 3 December 2015 in nonlinear optics with certain inhomogeneous medium existing. By virtue of a kind of the Availableonline12December2015 Darboux transformation, under the Painlevé integrable condition, the first- and second-order bright and dark rogue-wave solutions are derived. Properties of the first- and second-order Keywords: α β Inhomogeneous nonlinear system bright and dark rogue waves with (t), which measures the state of the basic flow, and (t), Baroclinic wave packets in geophysical fluids representing the interaction of the wave packet and mean flow, are graphically presented and Ultra-short pulses in nonlinear optics analyzed: α(t)andβ(t) have no influence on the wave packet, but affect the correction of the Rogue-wave solution basic flow. -

Nonlinear Optics in Daily Life
Nonlinear optics in daily life Elsa Garmire* Thayer School of Engineering, Dartmouth College, 14 Engineering Drive, Hanover, NH, 03755, USA *[email protected] Abstract: An overview is presented of the impact of NLO on today’s daily life. While NLO researchers have promised many applications, only a few have changed our lives so far. This paper categorizes applications of NLO into three areas: improving lasers, interaction with materials, and information technology. NLO provides: coherent light of different wavelengths; multi-photon absorption for plasma-materials interaction; advanced spectroscopy and materials analysis; and applications to communications and sensors. Applications in information processing and storage seem less mature. © 2013 Optical Society of America OCIS codes: (190.0190) Nonlinear optics; (060.0060) Fiber optics and optical communications; (120.0120) Instrumentation, measurement, and metrology; (140.0140) Lasers and laser optics; (180.0180) Microscopy; (300.0300) Spectroscopy. References and Links 1. E. Garmire, “Overview of nonlinear optics,” open source, found at http://www.intechopen.com/books/nonlinear- optics/overview-of-nonlinear-optics. 2. E. Garmire, “Resource letter NO-1: nonlinear optics,” Am. J. Phys. 79(3), 245–255 (2011). 3. http://en.wikipedia.org/wiki/Laser_pointer. 4. http://www.coherent.com/products/?921/Mira-Family. 5. C. G. Durfee, T. Storz, J. Garlick, S. Hill, J. A. Squier, M. Kirchner, G. Taft, K. Shea, H. Kapteyn, M. Murnane, and S. Backus, “Direct diode-pumped Kerr-lens mode-locked Ti:sapphire laser,” Opt. Express 20(13), 13677– 13683 (2012). 6. http://www.continuumlasers.com/products/tunable_horizon_OPO.asp. 7. http://www.newport.com/Inspire-Automated-Ultrafast-OPOs/847655/1033/info.aspx. -

Light-Wave Mixing and Scattering with Quantum Gases
23rd International Laser Physics Workshop (LPHYS’14) IOP Publishing Journal of Physics: Conference Series 594 (2015) 012011 doi:10.1088/1742-6596/594/1/012011 Light-wave mixing and scattering with quantum gases L. Deng and E.W. Hagley National Institute of Standards and Technology, Gaithersburg, Maryland 20899 USA C.J. Zhu School of Physical Science and Engineering, Tongji University, Shanghai 200092, China E-mail: [email protected] Abstract: We show that optical processes originating from elementary excitations with dominant collective atomic recoil motion in a quantum gas can profoundly change many nonlinear optical processes routinely observed in a normal gas. Not only multi-photon wave mixing processes all become stimulated Raman or hyper-Raman in nature but the usual forward wave-mixing process, which is the most efficient process in normal gases, is strongly reduced by the condensate structure factor. On the other hand, in the backward direction the Bogoliubov dispersion automatically compensates the optical- wave phase mismatch, resulting in efficient backward light field generation that usually is not supported in normal gases. 1. Introduction Since the invention of the laser in 1960, the field of nonlinear optics has exploded. In a typical nonlinear optical process, often referred to as coherent wave-mixing, several photons are absorbed simultaneously by an atomic or molecular medium, and a photon with a different frequency is produced. Under the right conditions, the generated radiation may be coherently amplified by the quantum mechanical mechanism of stimulated emission identified by Albert Einstein. Today, 95 years after Einstein’s prediction, we are surrounded by practical applications of nonlinear optics, from multi-channel telecom lasers to the common laser pointer. -

Learning Dominant Physical Processes with Data-Driven Balance Models ✉ Jared L
ARTICLE https://doi.org/10.1038/s41467-021-21331-z OPEN Learning dominant physical processes with data-driven balance models ✉ Jared L. Callaham 1 , James V. Koch2, Bingni W. Brunton 3, J. Nathan Kutz4 & Steven L. Brunton1 Throughout the history of science, physics-based modeling has relied on judiciously approximating observed dynamics as a balance between a few dominant processes. How- ever, this traditional approach is mathematically cumbersome and only applies in asymptotic 1234567890():,; regimes where there is a strict separation of scales in the physics. Here, we automate and generalize this approach to non-asymptotic regimes by introducing the idea of an equation space, in which different local balances appear as distinct subspace clusters. Unsupervised learning can then automatically identify regions where groups of terms may be neglected. We show that our data-driven balance models successfully delineate dominant balance physics in a much richer class of systems. In particular, this approach uncovers key mechanistic models in turbulence, combustion, nonlinear optics, geophysical fluids, and neuroscience. 1 Department of Mechanical Engineering, University of Washington, Seattle, WA, USA. 2 Oden Institute for Computational & Engineering Sciences, University of Texas, Austin, TX, USA. 3 Department of Biology, University of Washington, Seattle, WA, USA. 4 Department of Applied Mathematics, University of ✉ Washington, Seattle, WA, USA. email: [email protected] NATURE COMMUNICATIONS | (2021) 12:1016 | https://doi.org/10.1038/s41467-021-21331-z | www.nature.com/naturecommunications 1 ARTICLE NATURE COMMUNICATIONS | https://doi.org/10.1038/s41467-021-21331-z t is well known across the engineering and physical sciences such models is not always necessary to describe the local behavior Ithat persistent behaviors in complex systems are often deter- of a system. -

Nonlinear Optics and Nanophotonics November 25 - Decem Ber 6, 2013 Instituto De Física Teórica - UNESP - São Paulo, Brazil
ICTP International Centre for Theoretical Physics SAIFR South American Institute for Fundamental Research São Paulo International Schools on Theoretical Physics Nonlinear Optics and Nanophotonics November 25 - Decem ber 6, 2013 Instituto de Física Teórica - UNESP - São Paulo, Brazil This school on Nonlinear Optics and Nanophotonics will cover the physics of nonlinear optical systems including photonic crystals and photonic lattices; Nonlinear optics at the nanoscale; Active optical media and laser dynamics; Nonlinear effects in optical communication systems; Metamaterials; Complex dynamics of quantum optical systems. ICTP-SAIFR Steering Committee: The school will end with a one day Workshop and precede the 6th Rio de la Plata Fernando Quevedo (chair) Workshop on Laser Dynamics and Nonlinear Photonics, which will be held in Montevideo, ICTP director Uruguay from December 9-12, 2013 (http://www.fisica.edu.uy/~cris/workshop.htm). Julio Cezar Durigan UNESP rector Carlos Brito Cruz Lecturers: FAPESP scientific director Thorsten Ackemann: Laser solitons, VCSEL, patterns and dynamics Jacob Palis University of Strathclyde, Scotland, UK Brasil Acad. of Sci. president Juan Maldacena Cid de Araujo: Nonlinear optical processes in condensed matter Representing South America Universidade Federal de Pernambuco, Brazil ICTP-SAIFR Marcel Clerc: Spatiotemporal chaotic localized states in optics Scientific Council: Universidad de Chile Peter Goddard (chair) Alexander Gaeta: Nonlinear interactions in optical waveguides IAS, Princeton Cornell University, USA S. Randjbar-Daemi (coord.) ICTP vice-director Dan Gauthier: Tailoring the group velocity and group velocity dispersion Juan Montero Duke University, USA IFT-UNESP director Marcela Carena Michal Lipson: Nanophotonic structures for extreme nonlinearities on-chip Fermilab, Batavia Cornell University, USA Marcel Clerc Univ. -

L1 Logistics. Intro To
OSE 6334 Nonlinear Optics Konstantin Vodopyanov 1 1 Lecture 1 Introduction to nonlinear optics. Logistics. 2 2 1 What this course is about This course gives a broad introduction to the field of Nonlinear Optics (NLO). The goal is to get you acquainted with the main effects of nonlinear optics and also introduce main equations that govern nonlinear-optical interactions. Beginning with a simple electron on a spring model—the course gives comprehensive explanations of second-order and third-order nonlinear effects and their physical origins. The corse covers nonlinear optics from a combined: physics, optics, materials science, and devices perspective. Examples will be given on solving practical problems such as design of nonlinear frequency converters, and also devices using ultrafast and high intensity lasers. 3 3 Newton’s superposition principle f(A+B) = f(A) + f(B) B Isaac Newton 1642 –1726 A Newton: Light cannot interact with light. Two beams of light can have no effect on each other. 4 4 2 Nonlinear optics f(A+B) ≠ f(A) + f(B) Beams A and B can create beam C Nicolaas Bloembergen B 1920–2017 Nobel Prize in Physics (1981) C A NLO: photons do interact 5 5 Linear Optics • The optical properties such as the refractive index and the absorption coefficient are independent of light intensity. • The Newton’s principle of superposition: light cannot interact with light. Two beams of light in the same region of a linear optical medium can have no effect on each other. Thus light cannot control light. • The frequency of light cannot be altered by its passage through the medium. -

Entropic Thermodynamics of Nonlinear Photonic Chain Networks
ARTICLE https://doi.org/10.1038/s42005-020-00484-1 OPEN Entropic thermodynamics of nonlinear photonic chain networks Fan O. Wu 1,4, Pawel S. Jung1,2,4, Midya Parto 1, Mercedeh Khajavikhan3 & ✉ Demetrios N. Christodoulides 1 1234567890():,; The convoluted nonlinear behaviors of heavily multimode photonic structures have been recently the focus of considerable attention. The sheer complexity associated with such multimode systems, allows them to display a host of phenomena that are otherwise impossible in few-mode settings. At the same time, however, it introduces a set of funda- mental challenges in terms of comprehending and harnessing their response. Here, we develop an optical thermodynamic approach capable of describing the thermalization dynamics in large scale nonlinear photonic tight-binding networks. For this specific system, an optical Sackur-Tetrode equation is obtained that explicitly provides the optical tempera- ture and chemical potential of the photon gas. Processes like isentropic expansion/com- pression, Joule expansion, as well as aspects associated with beam cleaning/cooling and thermal conduction effects in such chain networks are discussed. Our results can be used to describe in an effortless manner the exceedingly complex dynamics of highly multimoded nonlinear bosonic systems. 1 CREOL, College of Optics and Photonics, University of Central Florida, Orlando, FL 32816-2700, USA. 2 Faculty of Physics, Warsaw University of Technology, Koszykowa 75, 00-662 Warsaw, Poland. 3 Department of Electrical and Computer Engineering, University of Southern California, Los Angeles, ✉ CA 9007, USA. 4These authors contributed equally: Fan O. Wu, Pawel S. Jung. email: [email protected] COMMUNICATIONS PHYSICS | (2020) 3:216 | https://doi.org/10.1038/s42005-020-00484-1 | www.nature.com/commsphys 1 ARTICLE COMMUNICATIONS PHYSICS | https://doi.org/10.1038/s42005-020-00484-1 n recent years, there has been considerable interest in available modes M, and the input optical power P. -

Laboratory of Physics and Modeling of Condensed Matter – Institut Néel Phd Thesis Project
Laboratory of Physics and Modeling of Condensed Matter – Institut Néel PhD thesis project Quantum and nonlinear optics of disordered media General context: Quantum communications and quantum information processing, including the rapidly developing field of quantum reservoir computing, often rely on optical qubits as information carriers. In this context, the robustness of the physical processes against decoherence induced by disordered media is of major importance at any steps: the photon production, propagation, processing, etc. especially in view of maintaining the quantum correlation properties of entangled photons. From a wider point of view, multiple light scattering in strongly disordered media (dense suspensions or powders of nanoparticles, clouds of cold atoms, etc.) is known to lead to numerous mesoscopic phenomena including Anderson localization, universal fluctuations of transmission (or “conductance”), long- and infinite-range correlation of scattered intensity, that strongly impact the quantum properties of light. Subject, available means: This project is a combined experimental and theoretical effort aimed at understanding quantum and nonlinear optical processes in the presence of disorder that induces strong multiple scattering of light. In a typical experiment, a powerful light pulse will be sent into a suspension or a powder of sub-micron-sized particles and the nonlinear signal generated by the medium (e.g., second harmonics or parametrically generated entangled photon pairs) will be measured and its quantum statistics will be studied. Another configuration to consider is that of an entangled photon pair is transmitted through a strongly scattering nonlinear medium for analyzing the persistence of entanglement with clear applications to information transfer through foggy air. Most of experimental means are already available (pulsed laser for SHG, Signac loop for entangled photons generation as well as the detection based on four photon correlation measurements).