Medical Genetics Summaries
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Power Gold: 2013’S Ins & Outs for the Second Consecutive Year, Country’S Top 100 Power Gold List Displays a Lot of Turnover from the Previous 12 Months
April 29, 2013, Issue 343 Power Gold: 2013’s Ins & Outs For the second consecutive year, Country’s Top 100 Power Gold list displays a lot of turnover from the previous 12 months. This year’s Top 100 boasts 22 new titles, compared to a whopping 33 in our April 2012 analysis and 13 new tunes in July 2011. This year’s Top 10 Power Gold is culled from Mediabase 24/7 and the Country Aircheck/Mediabase reporting panel for the week of April 21-27: 1. Kid Rock/All Summer Long 2. Zac Brown Band/Chicken Fried 3. Keith Urban/Somebody Like You 4. Alan Jackson & Jimmy Buffet/It’s Five O’Clock Somewhere 5. Dierks Bentley/What Was I Thinkin’ 6. Zac Brown Band/Toes 7. Rodney Atkins/Watching You Keith Urban Man, What Are You Doing Here? Rodeowave’s Phil Vassar (c) invites KSON/San Diego’s John & Tammy on stage while he 8. Rascal Flatts/Life Is A Highway plays “Piano Man” at the Stagecoach Country Music Festival in 9. Joe Nichols/Gimmie That Girl Indio, CA Saturday (4/27). 10. Rodney Atkins/If You’re Goin’ Through Hell New to the Top 10 are the ZBB, Flatts and Atkins tunes. Pope Finds Her Voice Sliding out of the Top 10 were Darius Rucker’s “Alright” She brought a pop-rock background and sound to The (16); Carrie Underwood’s “Before He Cheats” (15) and Voice’s Team Blake, but Cassadee Pope is now readying James Otto’s “Just Got Started Lovin’ You” (20). her Republic Nashville country debut. -

Layout 1 (Page 2)
AUGUST 12-18, 2011 CURRENTSURRENTS CThe News-Review’s guide to arts, entertainment and television TURBULENT TROUBADOUR Roseburg musician brings Texas-style blues to show Saturday MICHAEL SULLIVAN/The News-Review INSIDE: What’s Happening/3 Calendar/4 Galleries/6 Book Review/10 TV/15 Page 2, The News-Review Roseburg, Oregon, Currents—Thursday, August 11, 2011 * &YJUt$BOZPOWJMMF 03t*OGPt3FTtTFWFOGFBUIFSTDPN Roseburg, Oregon, Currents—Thursday, August 11, 2011 The News-Review, Page 3 what’s HAPPENING ROSEBURG tonight, blues artists Buddy Guy and Jimmie Vaughan Fri- Optimist hosts FOUR-SQUEAL DRIVE day and tribute band Beatle- Mania Live! Saturday. Con- band fundraiser certs on the Umpqua Park The Roseburg Optimist Club stage start at 8 p.m. each night. hosts its Battle of the Bands Daily, 4-H and FFA events fundraiser from 6 to 10 p.m. begin at 9 a.m., carnival gates Saturday at Pyrenees Vineyard open from 10 a.m. to 11 p.m. and Cellars, north location, daily, exhibit buildings open 707 Hess Lane, Roseburg. 11 a.m. to 11 p.m. Local musi- Music entertainment cal acts happen throughout the includes Not Penny’s Boat, day on the Charter Communi- The Dixonville Chicks and cations Stage. Dylan James. Reserved seating for con- Advance tickets are $20 certs is extra and tickets are each or $35 for a couple. Price available at the fair office or at the door is $25 per person. ticketsoregon.com. General Tickets include wine tasting seating is free with admission and sandwiches. to the fairgrounds. Information: 541-672-8060 Admission is $9 for adults or 13 years and older, $7 for sen- www.pyreneesvineyard.com. -
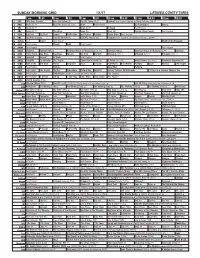
Sunday Morning Grid 1/1/17 Latimes.Com/Tv Times
SUNDAY MORNING GRID 1/1/17 LATIMES.COM/TV TIMES 7 am 7:30 8 am 8:30 9 am 9:30 10 am 10:30 11 am 11:30 12 pm 12:30 2 CBS CBS News Sunday Face the Nation (N) The NFL Today (N) Å Football New England Patriots at Miami Dolphins. (N) Å 4 NBC News (N) Å Meet the Press (N) (TVG) News Paid Program Oz Reimagined Hockey 5 CW News (N) Å News (N) Å In Touch Paid Program 7 ABC News (N) Å This Week News (N) News (N) Florida Citrus Parade Paid Program 9 KCAL Big Deal Big Deal People CHiP’s-Kids Joel Osteen Schuller Pastor Mike Paid Program 11 FOX Fox News Sunday FOX NFL Kickoff (N) FOX NFL Sunday (N) Football Dallas Cowboys at Philadelphia Eagles. (N) Å 13 MyNet Paid Matter Paid Program March of the Penguins 18 KSCI Paid Program Church Faith Paid Program 22 KWHY Paid Program Paid Program 24 KVCR Downton Downton Abbey Downton Abbey on Masterpiece (8:37) Downton Abbey Downton Abbey on Masterpiece Å Downton 28 KCET 1001 Nights Bug Bites Bug Bites Edisons Biz Kid$ Biz Kid$ Artbound (TVY) Å Artbound (TVY) Å Artbound (TVY) Å 30 ION Jeremiah Youssef In Touch Paid Program 34 KMEX Conexión En contacto Paid Program Como Dice el Dicho (N) Al Punto (N) (TVG) Netas Divinas (N) (TV14) República Deportiva (N) 40 KTBN Walk in the Win Walk Prince Carpenter Jesse In Touch PowerPoint It Is Written Pathway Super Kelinda John Hagee 46 KFTR Paid Program Película Zona NBA Película 50 KOCE Odd Squad Odd Squad Martha Cyberchase Clifford-Dog WordGirl Forever Painless With Miranda 21 Days to a Slimmer Younger You 52 KVEA Paid Program Fútbol Inglés Arsenal FC vs Crystal Palace FC. -
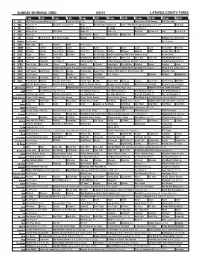
Sunday Morning Grid 8/2/15 Latimes.Com/Tv Times
SUNDAY MORNING GRID 8/2/15 LATIMES.COM/TV TIMES 7 am 7:30 8 am 8:30 9 am 9:30 10 am 10:30 11 am 11:30 12 pm 12:30 2 CBS CBS News Sunday Morning (N) Å Face the Nation (N) Paid Program Road to the PGA Champ. PGA Tour Golf 4 NBC News (N) Å Meet the Press (N) Å News Mecum Auto Auction (N) 2015 FINA World Championships (Taped) Å Red Bull 5 CW News (N) Å News (N) Å In Touch Paid Program 7 ABC News (N) Å This Week News (N) News (N) News Å Eye on L.A. Paid Eye on L.A. 9 KCAL News (N) Joel Osteen Hour Mike Webb Woodlands Paid Program 11 FOX In Touch Joel Osteen Fox News Sunday Midday Paid Program Hostage ›› (2005) (R) 13 MyNet Paid Program Paid Program 18 KSCI Man Land Paid Church Faith Paid Program 22 KWHY Cosas Contac Jesucristo Local Local Gebel Local Local Local Local RescueBot Transfor. 24 KVCR Painting Dowdle Joy of Paint Wyland’s Paint This Oil Painting Kitchen Mexican Cooking BBQ Simply Ming Lidia 28 KCET Raggs Space Travel-Kids Biz Kid$ News Asia Insight Soulful Symphony With Darin Atwater: Song Do the Math 30 ION Jeremiah Youssef In Touch Bucket-Dino Bucket-Dino Doki (TVY) Doki (TVY) Dive, Olly Dive, Olly The Karate Kid ››› 34 KMEX Paid Conexión Al Punto (N) Fútbol Central (N) Fútbol Mexicano Primera División: Toluca vs Pumas República Deportiva (N) 40 KTBN Walk in the Win Walk Prince Carpenter Hour of In Touch PowerPoint It Is Written Pathway Super Kelinda Jesse 46 KFTR Paid Program Dennis the Menace ›› (1993) Walter Matthau. -

DEATH VALLEY Credits Annie Gunn
Presents DEATH VALLEY Credits Annie Gunn..................................................Katrina Law Billy Rich.................................................Lochlyn Munro Jamie Dillen...............................................Victoria Pratt Roy Dillen...............................................Nick E. TaraBay Greenstreet.......................................................Kelly Hu Lucas Kern..........................................Jeremy Ratchford Holly Fields.............................................Juliette Beavan Lyla Dahl.........................................................Cela Scott Marco……………………………………………..Brad MacPherson Director.............................................................TJ Scott Written By........................TJ Scott & Brad MacPherson Executive Producer..................................Wence Sulda Producer..............................................Taylor Williams Producer..........................................Brad MacPherson Producer..........................................................TJ Scott Co-producer...........................................Jennie Yamaki Co-Producer...................................................Bert Kish First Assistant Director..........................Mark Wallace Second Assistant Director........Marie-Helene Riverain Original Music......................................................8MM Director of Photography...................David Herrington Unit Production manager……….…….......Jennie Yamaki Editor.............................................................Bert -

Steve Wennerstrom Papers Finding
T h e Steve Wennerstrom Papers The H.J. Lutcher Stark Center for Physical Culture & Sports The University of Texas at Austin Finding Aid Steve Wennerstrom Papers: 77 Boxes, 2 Oversized Boxes: 2833 Folders, 17 Artifacts, 130 Videos, no date, 1836-2014 (11491 items) Abstract Steve Wennerstrom is an expert on the history of women’s bodybuilding. He has served as the International Federation of Bodybuilders (IFBB) Women’s Historian. However, his interest in women’s sports extends beyond bodybuilding and the collection reflects that reality. Almost every sport that women have played is represented, making the Steve Wennerstrom Papers an invaluable resource to all researchers with an interest in women’s sports. Wennerstrom graduated from Cal State Fullerton and ran track. He also created one of the first women’s track publications, Women’s Track and Field World. The collection covers everything from strongwomen and circus performers to bodybuilding and powerlifting. It is truly an amazing knowledge base for bodybuilding and sports fans everywhere, especially those interested in the rise of women’s sports throughout the recent decades. Access Access to the Steve Wennerstrom Papers is restricted to visitors of the H.J. Lutcher Stark Center for Physical Culture and Sports and must be requested in writing prior to arrival at the Center. Furthermore, certain items in the collection (as identified below) are not suitable to be viewed by minors. The research request/proposal should explain the proposed project, the expected outcome, and institutional affiliation, if any. Requests should be sent to: Dr. Jan Todd at [email protected]. -

Amp 2019 Table of Contents
November 7-9, 2019 Baltimore Convention Center Baltimore, MD, USA AMP_OnsiteProgram_Covers.indd 1 10/18/19 9:39 AM PICKING UP PIK3CA WE SEE THE FUTURE OF GENOMIC MUTATIONS DIAGNOSTICS CLEARLY. Learn about the most common mutation in HR+/HER2- advanced breast cancer (aBC) 1-4 and how to detect it at BOOTH 2741 Personal Genome Diagnostics (PGDx) is Empowering the Fight Against Cancer by unlocking actionable information from the genome. We are committed to improving clinical insight, speed of results, and health economics by developing an INNOVATION SPOTLIGHT innovative portfolio of regulated with Jean Lopategui, MD Associate Professor of Pathology tissue-based and non-invasive liquid Director of Translational Genomics Program Director of Molecular Genetic biopsy genomic based Next Pathology Fellowship Generation Sequencing (NGS) Cedars-Sinai Medical Center Los Angeles, California products for laboratories worldwide. Friday, November 8, 2019 We are placing the power of proximity 10:00 AM - 10:30 AM in the hands of physicians and lab at STAGE 2 directors, and putting the power of control back into your patient care Join us as Dr Jean Lopategui discusses PIK3CA mutations in ecosystem. HR+/HER2- aBC and how to detect them. References: 1. The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61-70. 2. Tolaney S, Toi M, Neven P, et al. Presented at: DRIVEN BY SCIENCE. 2019 American Association for Cancer Research (AACR) Annual Meeting; March 29-April 3, 2019; INSPIRED BY PATIENTS. Atlanta, GA. 3. Di Leo A, Johnston S, Seok Lee K, et al. Lancet Oncol. -

Hopefuls Pitch Ideas for Tooele
www.tooeletranscript.com THURSDAY TOOELE Photo RANSCRIPT display T captures spirit of West, Mustangs See B1 BULLETIN September 29, 2005 SERVING TOOELE COUNTY SINCE 1894 VOL. 112 NO. 37 50 cents Not Playing With Fire Primary Elections 2005 Hopefuls pitch ideas for Tooele By Mark Watson Bulletin invited these can- STAFF WRITER didates to provide personal Six candidates are run- background information ning to fill two open posi- about why they would be tions on the Tooele City strong city leaders. Council. Voters will trim that John Hansen list of candidates to four at Hansen’s experience Tuesday’s primary election. includes service in various Doug Redmond will run to capacities including an Army retain his seat on the coun- veteran, councilman, Tooele cil. He will complete a four- Federal Credit Union direc- year term at the end of the tor, retired logistics director year. Also running is incum- Dugway Proving Ground, bent John Hansen. He was retired commanding general appointed to the council in of Utah State Defense Force, December 2004 when for- retired engineer group com- mer council member Colleen mander over all engineering Johnson vacated her seat forces in the Utah National photography / after being elected a Tooele Guard, former commander Troy Boman County Commissioner. of Army Garrison at Camp Tooele Also vying for the two Williams, former commander County open spots on the coun- of Utah Military Academy, employee cil are David McCall, Tom former Deputy Commander Diane Poyner, Scott Wardle and of 19th Special Forces Group Burgener Sam Woodruff. practices The Tooele Transcript- SEE COUNCIL ON A5 with a fire extinguisher as part of a safety training pro- Stockton candidates gram at the courthouse Thursday split on sewer plan morning. -

The Essential Science Fiction Television Reader
University of Kentucky UKnowledge American Popular Culture American Studies 5-2-2008 The Essential Science Fiction Television Reader J. P. Telotte Georgia Institute of Technology Click here to let us know how access to this document benefits ou.y Thanks to the University of Kentucky Libraries and the University Press of Kentucky, this book is freely available to current faculty, students, and staff at the University of Kentucky. Find other University of Kentucky Books at uknowledge.uky.edu/upk. For more information, please contact UKnowledge at [email protected]. Recommended Citation Telotte, J. P., "The Essential Science Fiction Television Reader" (2008). American Popular Culture. 8. https://uknowledge.uky.edu/upk_american_popular_culture/8 THE ESSENTIAL SCIENCE FICTION TELEVISION READER ESSENTIAL READERS IN CONTEMPORARY MEDIA AND CULTURE This series is designed to collect and publish the best scholarly writing on various aspects of television, film, the Internet, and other media of today. Along with providing original insights and explorations of criti- cal themes, the series is intended to provide readers with the best avail- able resources for an in-depth understanding of the fundamental issues in contemporary media and cultural studies. Topics in the series may include, but are not limited to, critical-cultural examinations of cre- ators, content, institutions, and audiences associated with the media industry. Written in a clear and accessible style, books in the series include both single-author works and edited collections.