Do You Know About Barth Syndrome? Eli (Age 2)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

SUPPLEMENTARY MATERIAL Effect of Next
SUPPLEMENTARY MATERIAL Effect of Next-Generation Exome Sequencing Depth for Discovery of Diagnostic Variants KKyung Kim1,2,3†, Moon-Woo Seong4†, Won-Hyong Chung3, Sung Sup Park4, Sangseob Leem1, Won Park5,6, Jihyun Kim1,2, KiYoung Lee1,2*‡, Rae Woong Park1,2* and Namshin Kim5,6** 1Department of Biomedical Informatics, Ajou University School of Medicine, Suwon 443-749, Korea 2Department of Biomedical Science, Graduate School, Ajou University, Suwon 443-749, Korea, 3Korean Bioinformation Center, Korea Research Institute of Bioscience and Biotechnology, Daejeon 305-806, Korea, 4Department of Laboratory Medicine, Seoul National University Hospital College of Medicine, Seoul 110-799, Korea, 5Department of Functional Genomics, Korea University of Science and Technology, Daejeon 305-806, Korea, 6Epigenomics Research Center, Genome Institute, Korea Research Institute of Bioscience and Biotechnology, Daejeon 305-806, Korea http//www. genominfo.org/src/sm/gni-13-31-s001.pdf Supplementary Table 1. List of diagnostic genes Gene Symbol Description Associated diseases ABCB11 ATP-binding cassette, sub-family B (MDR/TAP), member 11 Intrahepatic cholestasis ABCD1 ATP-binding cassette, sub-family D (ALD), member 1 Adrenoleukodystrophy ACVR1 Activin A receptor, type I Fibrodysplasia ossificans progressiva AGL Amylo-alpha-1, 6-glucosidase, 4-alpha-glucanotransferase Glycogen storage disease ALB Albumin Analbuminaemia APC Adenomatous polyposis coli Adenomatous polyposis coli APOE Apolipoprotein E Apolipoprotein E deficiency AR Androgen receptor Androgen insensitivity -

The Limb-Girdle Muscular Dystrophies and the Dystrophinopathies Review Article
Review Article 04/25/2018 on mAXWo3ZnzwrcFjDdvMDuzVysskaX4mZb8eYMgWVSPGPJOZ9l+mqFwgfuplwVY+jMyQlPQmIFeWtrhxj7jpeO+505hdQh14PDzV4LwkY42MCrzQCKIlw0d1O4YvrWMUvvHuYO4RRbviuuWR5DqyTbTk/icsrdbT0HfRYk7+ZAGvALtKGnuDXDohHaxFFu/7KNo26hIfzU/+BCy16w7w1bDw== by https://journals.lww.com/continuum from Downloaded Downloaded from Address correspondence to https://journals.lww.com/continuum Dr Stanley Jones P. Iyadurai, Ohio State University, Wexner The Limb-Girdle Medical Center, Department of Neurology, 395 W 12th Ave, Columbus, OH 43210, Muscular Dystrophies and [email protected]. Relationship Disclosure: by mAXWo3ZnzwrcFjDdvMDuzVysskaX4mZb8eYMgWVSPGPJOZ9l+mqFwgfuplwVY+jMyQlPQmIFeWtrhxj7jpeO+505hdQh14PDzV4LwkY42MCrzQCKIlw0d1O4YvrWMUvvHuYO4RRbviuuWR5DqyTbTk/icsrdbT0HfRYk7+ZAGvALtKGnuDXDohHaxFFu/7KNo26hIfzU/+BCy16w7w1bDw== Dr Iyadurai has received the Dystrophinopathies personal compensation for serving on the advisory boards of Allergan; Alnylam Stanley Jones P. Iyadurai, MSc, PhD, MD; John T. Kissel, MD, FAAN Pharmaceuticals, Inc; CSL Behring; and Pfizer, Inc. Dr Kissel has received personal ABSTRACT compensation for serving on a consulting board of AveXis, Purpose of Review: The classic approach to identifying and accurately diagnosing limb- Inc; as journal editor of Muscle girdle muscular dystrophies (LGMDs) relied heavily on phenotypic characterization and & Nerve; and as a consultant ancillary studies including muscle biopsy. Because of rapid advances in genetic sequencing for Novartis AG. Dr Kissel has received research/grant methodologies, -

Cardiomyopathy in a Male Patient with Neutropenia and Growth Delay
Folsi et al. Italian Journal of Pediatrics 2014, 40:45 http://www.ijponline.net/content/40/1/45 ITALIAN JOURNAL OF PEDIATRICS CASE REPORT Open Access Cardiomyopathy in a male patient with neutropenia and growth delay Veronica Folsi1, Nunzia Miglietti1, Annamaria Lombardi1, Sara Boccacci1, Tatiana Utyatnikova1, Chiara Donati1, Livia Squassabia1, Laura Gazzola1, Ilaria Bosio1, Adele Borghi2, Veronica Grassi3, Lucia D Notarangelo3 and Alessandro Plebani1,4* Abstract Neutropenia encompasses a family of neutropenic disorders, both permanent and intermittent, ranging from severe (<500 neutrophils/mm3) to mild (500–1500 neutrophils/mm3), which may also affect other organ systems such as the pancreas, central nervous system, heart, muscle and skin. Neutropenia can lead to life-threatening pyogenic infections whose severity is roughly inversely proportional to the circulating neutrophil counts. When neutropenia is detected, an attempt should be made to establish the etiology, and to distinguish acquired forms (the most frequent, including post viral neutropenia and autoimmune neutropenia) and congenital forms (rare disorders) that may be either isolated or part of a complex rare genetic disease. We report on a male patient initially diagnosed with isolated neutropenia who later turned out to be affected with Barth syndrome, a rare complex inherited disorder. Keywords: Barth syndrome, Neutropenia, Cardiomyopathy, Growth delay, Tafazzin gene Background that, when neutropenia is detected, patients should be Neutropenia is a laboratory finding associated with vari- carefully investigated for associated signs and symptoms able severity ranging from benign transient neutropenia, that may underlie a more complex and rare disorder. such as that due to mild viral infection, to congenital We report on a patient initially diagnosed with isolated neutropenia (CN). -

Barth Syndrome: a Life-Threatening Disorder Caused by Abnormal Cardiolipin Remodeling Vaishnavi Raja, Christian A
Raja V, Reynolds CA, Greenberg ML. J Rare Dis Res Treat. (2017) 2(2): 58-62 Journal of www.rarediseasesjournal.com Rare Diseases Research & Treatment Mini-review Open Access Barth syndrome: A life-threatening disorder caused by abnormal cardiolipin remodeling Vaishnavi Raja, Christian A. Reynolds, and Miriam L. Greenberg Department of Biological Sciences, Wayne State University, USA Article Info ABSTRACT Article Notes Barth syndrome (BTHS) is a rare X-linked genetic disorder characterized by Received: December 24, 2017 cardiomyopathy, skeletal myopathy, neutropenia, and organic aciduria. The Accepted: March 21, 2017 presence and severity of clinical manifestations are highly variable in BTHS, *Correspondence: even among patients with identical gene mutations. Currently, less than 200 Miriam L. Greenberg, Department of Biological Sciences, patients are diagnosed worldwide, but it is estimated that the disorder may Wayne State University, USA; be substantially under-diagnosed due to the variable spectrum of clinical E-mail: [email protected] manifestations. BTHS is caused by mutations in the gene tafazzin (TAZ), resulting in defective remodeling of cardiolipin (CL), the signature phospholipid © 2017 Miriam L. Greenberg. This article is distributed under of the mitochondrial membranes. Many of the clinical sequela associated with the terms of the Creative Commons Attribution 4.0 International License. BTHS can be directly attributed to mitochondria defects. In 2008, a definitive biochemical test was described based on detection of the abnormal CL profile characteristic of BTHS. This mini-review provides an overview of the etiology of BTHS, as well as a description of common clinical phenotypes associated with the disorder. Introduction Barth syndrome (BTHS) is a rare X-linked genetic disorder associated with a wide array of clinical manifestations, including cardiomyopathy, skeletal myopathy, neutropenia, 3-methylglutaconic aciduria, and hypercholesterolemia1-3. -

Barth Syndrome Journal Volume 11, Issue 2 Winter 2011
Barth Syndrome Journal www.barthsyndrome.org Volume 11, Issue 2 Winter 2011 Saving lives through education, advances in treatment, and fi nding a cure for Barth syndrome. Great News about Barth Syndrome Individuals' Longevity By Kate McCurdy, Board Member, Barth Syndrome Foundation or several years, the Barth Syndrome Foundation (BSF) has made the claim that individuals with Barth syndrome (BTHS) are living far longer than ever before. Now we are delighted Fto have the data to prove it. All of our collective efforts ARE making a difference! Photo courtesy of Amanda Clark ~ 2010 BSF was incorporated back in 2000, and by the end of that year, we had 41 living boys in our group who had been diagnosed with BTHS (see Graph #1 on page 4). As the literature indicated Inside this Issue then, most BTHS individuals died very young, and, indeed, you can see that fewer than 10% BTHS Longevity 1 / 4 of those we knew living then were over 15 years old. That year, however, the oldest BTHS 2012 Conference 1 / 5 individual in the world of whom we were aware was in his forties, and that gave us great hope. Sci/Med Sessions Letter from the Chairman 2 Compare these fi gures to the similar ones today (see Graph #2 on page 4). Our organization Letter from the 3 has grown to include 148 individuals from around the world living with a genetically confi rmed Executive Director diagnosis of BTHS. Now, more than 35% are older than 15 years old. Furthermore, the oldest Family Sessions 6 man living with BTHS whom we know of now is in his sixties! (Cont’d on page 4) 2012 Conference 7 Agenda Science Corner 8 A Continuum of Collaboration Sensory-Based Issues 9 & Barth Syndrome Barth Syndrome 2012 Scientific and Medical Sessions BTHS Library 10 Research Initiatives 11-12 By Matthew J. -

Pre-Implantation Genetic Diagnosis and Assisted Reproductive Technology in Haemophilia
PRE-IMPLANTATION GENETIC DIAGNOSIS AND ASSISTED REPRODUCTIVE TECHNOLOGY IN HAEMOPHILIA DR PENELOPE FOSTER MELBOURNE IVF WHAT IS PGD ? early embryo diagnosis allows selection of unaffected embryos for transfer to patient alternative to antenatal testing and termination of affected pregnancy MELBOURNE IVF TECHNIQUE OF PGD standard IVF cycle biopsy of 1 or 2 cells from day 3 embryo diagnostic testing on biopsied cells selection of embryos for transfer MELBOURNE IVF PGD IN HAEMOPHILIA OPTIONS Sex selection: if affected husband, all male offspring unaffected, all females carriers = select male embryos for transfer if carrier wife, 1/2 males affected, 1/2 females carrier = select female embryos for transfer MELBOURNE IVF PGD IN HAEMOPHILIA OPTIONS Specific gene detection avoids discarding unaffected male embryos avoids transfer of carrier female embryos MELBOURNE IVF SEX SELECTION - FISH FLUORESCENT IN-SITU HYBRIDISATION detects chromosome number cell from embryo fixed to slide apply FISH probes DNA sequences complementary to small segment of particular chromosome probes labelled with coloured fluorochromes coloured spots indicate presence of sequence 8-probe FISH – chromosomes 4,13,16,18,21,22,X,Y select euploid XX or XY embryos for transfer MELBOURNE IVF FISH ON BLASTOMERES 13, 16, 18, 21, 22 X, Y, 4 MELBOURNE IVF XX XY MELBOURNE IVF PGD FOR SPECIFIC GENE DETECTION DNA amplification by PCR 2 cells from embryo fragment analysis on DNA sequencer inclusion of informative markers individualised tests for each couple significant -

CARDIOMAN): a Randomised Placebo Controlled Pilot Trial Conducted by the Nationally Commissioned Barth Syndrome Service CARDIOMAN
Treatment of Barth Syndrome by CARDIOlipin MANipulation (CARDIOMAN): A randomised placebo controlled pilot trial conducted by the nationally commissioned Barth Syndrome Service CARDIOMAN IRAS No: 170371 ISRCTN No: EUDRACT no.: 2015-001382-10 R&I No: CS/2015/4775 Details of Sponsor: University Hospitals Bristol NHS Foundation Trust Research and Innovation Level 3, Education & Research Centre Upper Maudlin Street Bristol BS2 8AE Tel: 0117 342 0233 Fax: 0117 342 0239 Chief Investigators & Research Team Contact Details Chief Investigator Emeritus Professor of Paediatric Stem Cell Dr Guido Pieles Transplantation Professor Colin Graham Steward Consultant Paediatric Cardiologist Bristol Royal Hospital for Children Professor Colin G Steward Paul O’Gorman Building School of Cellular & Molecular Medicine Bristol Biomedical Sciences Building BS2 8BJ University of Bristol University Walk Tel: 0117 342 8296 Bristol, BS8 1TD Email: [email protected] Tel: 0117 342 8044 [email protected] Professor in Diabetes and Metabolic Professor of Epidemiology Endocrinology Professor Andrew Robert Ness Professor Julian Paul Hamilton-Shield School of Oral & Dental Sciences School of Clinical Sciences Biomedical Research Unit Offices Level 6 University Hospitals Bristol Education Centre Education Centre Lower Maudlin Street University Hospitals Bristol NHS Foundation Bristol BS1 2LY Trust Upper Maudlin Street Tel: (0117) 34 21751 Bristol, BS2 8AE Email: [email protected] Tel: 0117 342 0159 CARDIOMAN 12 February 2019 Protocol – version -

National Center for Advancing Translational Sciences 2014 Report
National Center for Advancing Translational Sciences 2014 REPORT NCATS’ Definitions of Translation and Translational Science: Translation is the process of turning observations in the laboratory, clinic and community into interventions that improve the health of individuals and the public — from diagnostics and therapeutics to medical procedures and behavioral changes. Translational science is the field of investigation focused on understanding the scientific and operational principles underlying each step of the translational process. NCATS studies translation on a system-wide level as a scientific and operational problem. Table of Contents Director’s Message . 1 Pre-Clinical Innovation . 3 Improving the Drug Development Process . 4 Assay Development and Screening Technology . 4 NCATS Chemical Genomics Center . 5 Bridging Interventional Development Gaps (BrIDGs) . 6 Repurposing Drugs . 7 NCATS Pharmaceutical Collection (NPC) . 7 New Therapeutic Uses . 8 Testing and Predictive Models . 10 Tissue Chip for Drug Screening . 10 Toxicology in the 21st Century (Tox21) . .11 RNA Interference (RNAi) . 13 Clinical Innovation . 14 Clinical and Translational Science Awards (CTSA) Program . 14 Accelerating Rare Diseases Research . 16 SM NIH/NCATS Global Rare Diseases Patient Registry Data Repository/GRDR . 16 Rare Diseases Clinical Research Network (RDCRN) . 17 Therapeutics for Rare and Neglected Diseases (TRND) . 18 Patient/Community Engagement and Health Information . 19 Partnerships and Collaborations . 22 Resources for Small Businesses . 24 Tools for Accelerating Translational Science . 25 Enabling Public Access to Data . 25 Appendix: Therapeutics for Rare and Neglected Diseases (TRND) Program 2013–2014 Annual Reports . 27 Front cover, left: The Biorepository at the Einstein-Montefiore Institute for Clinical and Translational Research is a quality-assured facility for acquisition, processing, storage and secure distribution of specimens with patient-specific annotations from the electronic medical record (Courtesy of Albert Einstein College of Medicine). -

TAFAZZIN Gene Tafazzin
TAFAZZIN gene tafazzin Normal Function The TAFAZZIN gene provides instructions for producing a protein called tafazzin. Several different versions (isoforms) of the tafazzin protein are produced from the TAFAZZIN gene. Most isoforms are found in all tissues, but some are found only in certain types of cells. The tafazzin protein is located in structures called mitochondria, which are the energy-producing centers of cells. Tafazzin is involved in altering a fat ( lipid) called cardiolipin, which plays critical roles in the mitochondrial inner membrane. The tafazzin protein adds a fatty acid called linoleic acid to the cardiolipin molecule, which enables cardiolipin to perform its functions. Cardiolipin is necessary for maintaining mitochondrial shape, energy production, and protein transport within cells. Health Conditions Related to Genetic Changes Barth syndrome More than 130 mutations in the TAFAZZIN gene have been found to cause Barth syndrome. This rare condition occurs almost exclusively in males and is characterized by a weakened heart (cardiomyopathy), muscle weakness, recurrent infections, and short stature. TAFAZZIN gene mutations that cause Barth syndrome result in the production of tafazzin proteins with little or no function. As a result, linoleic acid is not added to cardiolipin, which results in a reduction of functional cardiolipin. In addition, a variant of cardiolipin called monolysocardiolipin (MLCL) is formed. A lack of functional cardiolipin and an excess of MLCL are thought to cause problems with normal mitochondrial shape and functions such as energy production and protein transport. Tissues with high energy demands, such as the heart and other muscles, are most susceptible to cell death due to reduced energy production in mitochondria. -

Inherited Neutropenia Gene Sequencing Panel
Inherited Neutropenia Gene Sequencing Panel Genes Tested: against overwhelming bacterial infection. Nonsense and frameshift variants affecting the carboxyl terminus are AK2 AP3B1 CD40LG CLPB quite common, while missense variants are seen more CSF3R CXCR2 CXCR4 DNAJC21 commonly in cyclic neutropenia patients. However, there EFL1 EIF2AK3 ELANE G6PC3 is considerable overlap of genotype with phenotype, GATA1 GATA2 GFI1 HAX1 even within families. Mutations in GFI1, HAX1, G6PC3, VPS45 and CFS3R are much less frequent causes of HYOU1 JAGN1 LAMTOR2 LYST severe congenital neutropenia. Variants within the Cdc42 MRTFA RAB27A RAC2 RMRP binding site of WAS are also associated with an X-linked (MKL1) form of congenital neutropenia. RUNX1 SBDS SLC37A4 SMARCD2 The diagnostic criteria for SCN include: SRP54 STK4 TAZ TCIRG1 TCN2 TP53 USB1 VPS13B • Early childhood onset of profound neutropenia (<0.5 × 109/L) VPS45 WAS WDR1 WIPF1 • Recurrent life-threatening bacterial infections • Promyelocytic maturation arrest in the bone marrow Description: This panel detects the most common genetic causes Syndromic neutropenia: Significant neutropenia is a of severe congenital neutropenia as well as genetic common feature of several genetic syndromes associated syndromes often associated with neutropenia, with extra-hematopoietic abnormalities including Barth including Barth syndrome, Chediak-Higashi syndrome, syndrome, Cohen syndrome secondary to VPS13B variants, Clericuzio-type poikiloderma with neutropenia, Cohen Chediak-Higashi syndrome, Clericuzio-type poikiloderma syndrome, GATA1-related X linked cytopenia, GATA2 with neutropenia, GATA1-related X linked cytopenia, deficiency, glycogen storage disease type 1B, Griscelli GATA2 deficiency, glycogen storage disease type 1B, syndrome type 2, Hermansky-Pudlak syndrome type Griscelli syndrome type 2, Hermansky-Pudlak syndrome 2, p14 deficiency, Shwachman-Diamond syndrome, type 2, p14 deficiency, Shwachman-Diamond syndrome and WHIM syndrome, and Wiscott-Aldrich syndrome. -

Geneticsreferral Guidance Sheet Qualifying Genetics Conditions
Genetics Referral Guidance Sheet To qualify for a wish, a child must meet these criteria at the time of referral: 1. Older than 2½ years and younger than 18 years 2. Has not received a wish from another wish-granting organization 3. Diagnosed with a critical illness (i.e. a progressive, degenerative or malignant condition) that, despite adherence to the treatment plan, is currently placing the child’s life in jeopardy 4. Qualifying Genetics Conditions: Aicardi Goutieres disease Metachromatic leukodystrophy Alexander disease (symptomatic) Mucopolysaccharidosis disorders (symptomatic) Barth syndrome o Hunter syndrome Congenital anomaly, chromosomal or o Hurler disease single-gene condition with associated o Sanfilippo syndrome life-threatening complications such as: Niemann-Pick disease o Intractable/refractory/treatment-resistant Peroxisomal disorder seizures Pyruvate dehydrogenase deficiency with o Structural upper airway abnormalities or progression of disease chronic pulmonary symptoms Skeletal dysplasias or dysostosis with o Heart anomalies meeting cardiology chronic or degenerative pulmonary criteria complications o Kidney dysfunction meeting nephrology Sphingolipidosis with symptoms in the criteria pediatric period o Associated major gastrointestinal o Tay-Sachs dysfunctions leading to TPN dependency o GM1 gangliosidosis Gaucher disease, type II Trisomy 13 Infantile Pompe Trisomy 18 Krabbe disease Urea cycle (CPS, OTC, citrullinemia) and Leigh disease with MRI changes organic acidemia disorders (methylmalonic and propionic) with a history of Lesch-Nyhan syndrome hyperammonemic event Maple syrup urine disease with a history X-linked adrenal leukodystrophy with of hyperuremic event adrenal or brain findings There are other conditions that may be eligible for a wish when the condition includes life-threatening comorbidities that are currently placing the child’s life in jeopardy. -
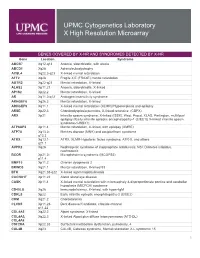
Genes Covered and Disorders Detected by X-HR Microarray
UPMC Cytogenetics Laboratory X High Resolution Microarray GENES COVERED BY X-HR AND SYNDROMES DETECTED BY X-HR Gene Location Syndrome ABCB7 Xq12-q13 Anemia, sideroblastic, with ataxia ABCD1 Xq28 Adrenoleukodystrophy ACSL4 Xq22.3-q23 X-linked mental retardation AFF2 Xq28 Fragile X E (FRAXE) mental retardation AGTR2 Xq22-q23 Mental retardation, X-linked ALAS2 Xp11.21 Anemia, sideroblastic, X-linked AP1S2 Xp22.2 Mental retardation, X-linked AR Xq11.2-q12 Androgen insensitivity syndrome ARHGEF6 Xq26.3 Mental retardation, X-linked ARHGEF9 Xq11.1 X-linked mental retardation (XLMR)//Hyperekplexia and epilepsy ARSE Xp22.3 Chondrodysplasia punctata, X-linked recessive (CDPX) ARX Xp21 Infantile spasm syndrome, X-linked (ISSX), West, Proud, XLAG, Partington, multifocal epilepsy //Early infantile epileptic encephalopathy-1 (EIEE1)( X-linked infantile spasm syndrome-1-ISSX1) ATP6AP2 Xp11.4 Mental retardation, X-linked, with epilepsy (XMRE) ATP7A Xq13.2- Menkes disease (MNK) and occipital horn syndrome q13.3 ATRX Xq13.1- ATRX, XLMR-Hypotonic facies syndrome, ATR-X, and others q21.1 AVPR2 Xq28 Nephrogenic syndrome of inappropriate antidiuresis; NSI; Diabetes insipidus, nephrogenic BCOR Xp21.2- Microphthalmia syndromic (MCOPS2) p11.4 BMP15 Xp11.2 Ovarian dysgenesis 2 BRWD3 Xq21.1 Mental retardation, X-linked 93 BTK Xq21.33-q22 X-linked agammaglobulinemia CACNA1F Xp11.23 Aland Island eye disease CASK Xp11.4 X-linked mental retardation with microcephaly & disproportionate pontine and cerebellar hypoplasia (MICPCH) syndrome CD40LG Xq26 Immunodeficiency,