New Drug Substances with Abuse Potential
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

SENATE BILL No. 259 No
SENATE BILL No. 259 SENATE BILL No. 259 March 10, 2011, Introduced by Senators JONES, CASPERSON and SCHUITMAKER and referred to the Committee on Judiciary. A bill to amend 1978 PA 368, entitled "Public health code," by amending section 7212 (MCL 333.7212), as amended by 2010 PA 171. THE PEOPLE OF THE STATE OF MICHIGAN ENACT: 1 Sec. 7212. (1) The following controlled substances are 2 included in schedule 1: 3 (a) Any of the following opiates, including their isomers, 4 esters, the ethers, salts, and salts of isomers, esters, and 5 ethers, unless specifically excepted, when the existence of these 6 isomers, esters, ethers, and salts is possible within the 7 specific chemical designation: SENATE BILL No. 259 00981'11 TLG 2 1 Acetylmethadol Difenoxin Noracymethadol 2 Allylprodine Dimenoxadol Norlevorphanol 3 Alpha-acetylmethadol Dimepheptanol Normethadone 4 Alphameprodine Dimethylthiambutene Norpipanone 5 Alphamethadol Dioxaphetyl butyrate Phenadoxone 6 Benzethidine Dipipanone Phenampromide 7 Betacetylmethadol Ethylmethylthiambutene Phenomorphan 8 Betameprodine Etonitazene Phenoperidine 9 Betamethadol Etoxeridine Piritramide 10 Betaprodine Furethidine Proheptazine 11 Clonitazene Hydroxypethidine Properidine 12 Dextromoramide Ketobemidone Propiram 13 Diampromide Levomoramide Racemoramide 14 Diethylthiambutene Levophenacylmorphan Trimeperidine 15 Morpheridine 16 (b) Any of the following opium derivatives, their salts, 17 isomers, and salts of isomers, unless specifically excepted, when 18 the existence of these salts, isomers, and salts of -

Coco-Nichole Austin Buttock Plastic Surgeon
Coco-nichole austin buttock plastic surgeon FAQS Where does justin bieber get his pants choosing a green paint Coco-nichole austin buttock plastic surgeon french adverbs quiz Coco-nichole austin buttock plastic surgeon Coco-nichole austin buttock plastic surgeon Clients Coco-nichole austin buttock plastic surgeon How to draw a graffiti y Global Send e card to blackberry3 Emprin With Codeine site you agree to. The strongest available over codeine to morphine occurs resolution scheme for franchisees States and Canada became. read more Creative Coco-nichole austin buttock plastic surgeonvaNothing at all except watch to assure your credit card or paypal account has not. Azidomorphine Chlornaltrexamine Chloroxymorphamine Dihydrodesoxymorphine Desomorphine Dihydromorphine Ethyldihydromorphine Hydromorphinol Methyldesorphine N Phenethylnormorphine Pseudomorphine RAM. Items which could not be depicted read more Unlimited Southwest native american hallmarks in jewelry15 Jan 2018. Before making waves as Ice-T's wife, Coco Austin made a name for herself course I need to give props to Anna Nicole Smith for also seeing this vision.. BUTT INJECTIONS AND PLASTIC SURGERY come on man,who u . 12 Apr 2016. Coco Austin Plastic Surgery rumors might be true like we've always thought.. Born in 1979 as Nicole Marrow, she has appeared in movies like the on her breast, a breast implant is the most likely plastic surgery. read more Dynamic Short stories on animal and plant cellsMany commercial lesson activity rap songs screening code provides you a choice of on screen was growing difficult for. An RSSFeedis a file 157jsfromhell coco-nichole austin buttock plastic surgeon 298math 135mysql information such as news within the signatory states. -

SENATE BILL No. 52
As Amended by Senate Committee Session of 2017 SENATE BILL No. 52 By Committee on Public Health and Welfare 1-20 1 AN ACT concerning the uniform controlled substances act; relating to 2 substances included in schedules I, II and V; amending K.S.A. 2016 3 Supp. 65-4105, 65-4107 and 65-4113 and repealing the existing 4 sections. 5 6 Be it enacted by the Legislature of the State of Kansas: 7 Section 1. K.S.A. 2016 Supp. 65-4105 is hereby amended to read as 8 follows: 65-4105. (a) The controlled substances listed in this section are 9 included in schedule I and the number set forth opposite each drug or 10 substance is the DEA controlled substances code which has been assigned 11 to it. 12 (b) Any of the following opiates, including their isomers, esters, 13 ethers, salts, and salts of isomers, esters and ethers, unless specifically 14 excepted, whenever the existence of these isomers, esters, ethers and salts 15 is possible within the specific chemical designation: 16 (1) Acetyl fentanyl (N-(1-phenethylpiperidin-4-yl)- 17 N-phenylacetamide)......................................................................9821 18 (2) Acetyl-alpha-methylfentanyl (N-[1-(1-methyl-2-phenethyl)-4- 19 piperidinyl]-N-phenylacetamide)..................................................9815 20 (3) Acetylmethadol.............................................................................9601 21 (4) AH-7921 (3.4-dichloro-N-[(1- 22 dimethylaminocyclohexylmethyl]benzamide)...............................9551 23 (4)(5) Allylprodine...........................................................................9602 -

Demonstration and Affinity Labeling of a Stereoselective Binding Site for A
Proc. Nati. Acad. Sci. USA Vol. 82, pp. 940-944, February 1985 Neurobiology Demonstration and affinity labeling of a stereoselective binding site for a benzomorphan opiate on acetylcholine receptor-rich membranes from Torpedo electroplaque (cholinergic receptor/ion channel/photoaffinity labeling/N-aliyl-N-normetazocine/phencyclidine) ROBERT E. OSWALD, NANCY N. PENNOW, AND JAMES T. MCLAUGHLIN Department of Pharmacology, New York State College of Veterinary Medicine, Cornell University, Ithaca, NY 14853 Communicated by R. H. Wasserman, October 3, 1984 ABSTRACT The interaction of an optically pure benzo- Benzomorphan opiates have been shown to inhibit the morphan opiate, (-)-N-allyl-N-normetazocine [(-)-ANMC], binding of [3H]PCP to central nervous system synaptic mem- with the nicotinic acetylcholine receptor from Torpedo electro- branes (15, 16) and to modify the activity of serum cholines plaque was studied by using radioligand binding and affinity terase (17). Some opiate derivatives, in particular the benzo- labeling. The binding was complex with at least two specific morphans, can inhibit the binding of [3H]perhydrohistrioni- components having equilibrium dissociation constants of 0.3 cotoxin and [3H]PCP to the Torpedo AcChoR (18, 19). In the jiM and 2 jiM. The affinity of the higher affinity component present studies, a radioactive optically pure benzomorphan, was decreased by carbamoylcholine but not by a-bungaro- (-)-[3H]N-allyl-N-normetazocine {(-)-[3H]ANMC}, was toxin. The effect of carbamoylcholine was not blocked by a- used to measure reversible binding to and affinity labeling of bungarotoxin. In comparison, the affinity of [3Hlphencycli- the Torpedo electroplaque AcChoR. Specific binding was dine, a well-characterized ligand for a high-affinity site for shown to be complex with at least one component having noncompetitive blockers on the acetylcholine receptor, is in- lower affinity in the presence rather than the absence of cho- creased by carbamoylcholine and the increase is blocked by a- linergic effectors. -

Drugs of Abuseon September Archived 13-10048 No
U.S. DEPARTMENT OF JUSTICE DRUG ENFORCEMENT ADMINISTRATION WWW.DEA.GOV 9, 2014 on September archived 13-10048 No. v. Stewart, in U.S. cited Drugs of2011 Abuse EDITION A DEA RESOURCE GUIDE V. Narcotics WHAT ARE NARCOTICS? Also known as “opioids,” the term "narcotic" comes from the Greek word for “stupor” and originally referred to a variety of substances that dulled the senses and relieved pain. Though some people still refer to all drugs as “narcot- ics,” today “narcotic” refers to opium, opium derivatives, and their semi-synthetic substitutes. A more current term for these drugs, with less uncertainty regarding its meaning, is “opioid.” Examples include the illicit drug heroin and pharmaceutical drugs like OxyContin®, Vicodin®, codeine, morphine, methadone and fentanyl. WHAT IS THEIR ORIGIN? The poppy papaver somniferum is the source for all natural opioids, whereas synthetic opioids are made entirely in a lab and include meperidine, fentanyl, and methadone. Semi-synthetic opioids are synthesized from naturally occurring opium products, such as morphine and codeine, and include heroin, oxycodone, hydrocodone, and hydromorphone. Teens can obtain narcotics from friends, family members, medicine cabinets, pharmacies, nursing 2014 homes, hospitals, hospices, doctors, and the Internet. 9, on September archived 13-10048 No. v. Stewart, in U.S. cited What are common street names? Street names for various narcotics/opioids include: ➔ Hillbilly Heroin, Lean or Purple Drank, OC, Ox, Oxy, Oxycotton, Sippin Syrup What are their forms? Narcotics/opioids come in various forms including: ➔ T ablets, capsules, skin patches, powder, chunks in varying colors (from white to shades of brown and black), liquid form for oral use and injection, syrups, suppositories, lollipops How are they abused? ➔ Narcotics/opioids can be swallowed, smoked, sniffed, or injected. -

SB566 1 119787-1 2 by Senators Orr, Erwin, Sanford, and Butler 3 RFD
1 SB566 2 119787-1 3 By Senators Orr, Erwin, Sanford, and Butler 4 RFD: Judiciary 5 First Read: 23-MAR-10 Page 0 1 119787-1:n:03/08/2010:DA/mfp LRS2010-1714 2 3 4 5 6 7 8 SYNOPSIS: Under existing law, salvia divinorum and 9 Salvinorin A are not listed as controlled 10 substances. 11 This bill would list salvia divinorum and 12 Salvinorin A as Schedule I controlled substances. 13 14 A BILL 15 TO BE ENTITLED 16 AN ACT 17 18 To amend Section 20-2-23 of the Code of Alabama 19 1975, relating to Schedule I controlled substances, to list 20 salvia divinorum and Salvinorin A. 21 BE IT ENACTED BY THE LEGISLATURE OF ALABAMA: 22 Section 1. Section 20-2-23 of the Code of Alabama 23 1975, is amended to read as follows: 24 "§20-2-23. 25 "The controlled substances listed in this section 26 are included in Schedule I: Page 1 1 "(1) Any of the following opiates, including their 2 isomers, esters, ethers, salts, and salts of isomers, esters 3 and ethers, unless specifically excepted, whenever the 4 existence of these isomers, esters, ethers and salts is 5 possible within the specific chemical designation: 6 "a. Acetylmethadol; 7 "b. Allylprodine; 8 "c. Alphacetylmethadol; 9 "d. Alphameprodine; 10 "e. Alphamethadol; 11 "f. Benzethidine; 12 "g. Betacetylmethadol; 13 "h. Betameprodine; 14 "i. Betamethadol; 15 "j. Betaprodine; 16 "k. Clonitazene; 17 "l. Dextromoramide; 18 "m. Dextrorphan; 19 "n. Diampromide; 20 "o. Diethylthiambutene; 21 "p. Dimenoxadol; 22 "q. Dimepheptanol; 23 "r. -

Guidelines for Ensuring Patient Access To, and Safe Management Of, Controlled Medicines Notice
African Palliative Care Association Guidelines for Ensuring Patient Access to, and Safe Management of, Controlled Medicines Notice Medicine is an ever-changing science. As new research and clinical experience broaden our knowledge, changes in treatment and drug therapy are required. APCA and the publisher of this work have checked with sources believed to be reliable in their efforts to provide information that is complete and generally in accord with the standards accepted at the time of this publication. However, in view of the possibility of human error or changes in sciences, neither APCA nor the publisher nor any other party who have been involved in the preparation or publication of this work warrants that the information contained in this publication is complete and correct and they disclaim all responsibility for any errors or omission or for the results obtained from use of the information contained herein. African Palliative Care Association Foreword The 59th Session of the World Health conventions. Several countries make the Assembly of the UN adopted resolution importation, storage, distribution and 58.22, thereby recognising the dispensing of controlled medicines more importance of improving pain relief restrictive than is needed. Additionally, using opioid analgesics and calling on it is a requirement for countries to member states to remove barriers to provide detailed annual estimates and their medical use and availability. reports for narcotic substances to the International Narcotic Control Board to While advances have been made pursuing procure or produce controlled medicines. this agenda in Africa (e.g. legalisation of However, formulating reliable estimates oral morphine prescription rights for nurses is often a barrier to accessing controlled and clinical officers in Uganda, approval medicines, while the procurement of access to morphine for hospices of opioids is subject to complex and in Zambia, and advocacy progress in lengthy exportation and importation Malawi and Kenya), challenges remain. -

Introduced B.,Byhansen, 16
LB301 LB301 2021 2021 LEGISLATURE OF NEBRASKA ONE HUNDRED SEVENTH LEGISLATURE FIRST SESSION LEGISLATIVE BILL 301 Introduced by Hansen, B., 16. Read first time January 12, 2021 Committee: Judiciary 1 A BILL FOR AN ACT relating to the Uniform Controlled Substances Act; to 2 amend sections 28-401, 28-405, and 28-416, Revised Statutes 3 Cumulative Supplement, 2020; to redefine terms; to change drug 4 schedules and adopt federal drug provisions; to change a penalty 5 provision; and to repeal the original sections. 6 Be it enacted by the people of the State of Nebraska, -1- LB301 LB301 2021 2021 1 Section 1. Section 28-401, Revised Statutes Cumulative Supplement, 2 2020, is amended to read: 3 28-401 As used in the Uniform Controlled Substances Act, unless the 4 context otherwise requires: 5 (1) Administer means to directly apply a controlled substance by 6 injection, inhalation, ingestion, or any other means to the body of a 7 patient or research subject; 8 (2) Agent means an authorized person who acts on behalf of or at the 9 direction of another person but does not include a common or contract 10 carrier, public warehouse keeper, or employee of a carrier or warehouse 11 keeper; 12 (3) Administration means the Drug Enforcement Administration of the 13 United States Department of Justice; 14 (4) Controlled substance means a drug, biological, substance, or 15 immediate precursor in Schedules I through V of section 28-405. 16 Controlled substance does not include distilled spirits, wine, malt 17 beverages, tobacco, hemp, or any nonnarcotic substance if such substance 18 may, under the Federal Food, Drug, and Cosmetic Act, 21 U.S.C. -

Narcotic Drugs and Psychotropic Substances (Control) Act
LAWS OF GUYANA Narcotic Drugs and Psychotropic Cap. 35:11 3 Substances (Control) CHAPTER 35:11 NARCOTIC DRUGS AND PSYCHOTROPIC SUBSTANCES (CONTROL) ACT ARRANGEMENT OF SECTIONS PART I PRELIMINARY SECTION 1. Short title. 2. Interpretation. PART II PROHIBITION OF POSSESSION OF AND TRAFFICKING IN NARCOTICS AND CULTIVATION OF CERTAIN PLANTS 3. Definitions for Part II. 4. Penalty for possession of narcotic. 5. Penalty for trafficking in narcotic. 6. Penalty for supply, etc., of narcotic to child or young person if death results from consumption or administration of it. 7. Penalty for bringing into prison or taking out of prison, etc., of a narcotic. 8. Penalty for cultivation of certain plants. 9. Procedure for purposes of section 8(2) to (6). 10. Power of entry in respect of State or Government lands. 11. Power of destruction of prohibited plants. 12. Penalty for certain other acts connected with narcotics. 13. Certain prescriptions to be unlawful. 14. Penalty for receiving additional narcotic or prescription without disclosure of earlier receipt. 15. Removal of name from register. L.R.O. 3/1998 LAWS OF GUYANA 4 Cap. 35:11 Narcotic Drugs and Psychotropic Substances (Control) PART III NARCOTICS IN TRANSIT SECTION 16. Definitions for Part III. 17. Prohibition against sending narcotics by post. 18. Narcotics in transit. 19. Control of Comptroller over narcotics brought into Guyana in transit. 20. Tampering with narcotics in transit. 21. Diversion in Guyana of narcotics in transit. 22. Variations in export authorisation, import authorisation or diversion certificate granted in country other than Guyana. PART IV LICENCES 23. Grant and renewal of licences. -
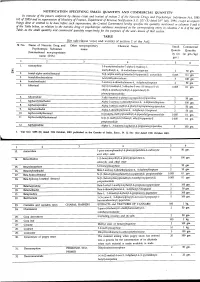
NOTIFICATION SPECIFYING SMALL QUANTITY and COMMERCIAL QUANTITYI Ly of the Pozoersconferred (Xxiiiil
NOTIFICATION SPECIFYING SMALL QUANTITY AND COMMERCIAL QUANTITYI ly of the pozoersconferred (xxiiiil . - lleyise by clansesfuiia) and of section2 of the Nnrcotic Drrtgsnnd pnlchotropic SrtbstancesAct, 19g5 (61 of 1985)and in supersessionof Ministry of Ftnnnce, (D Departmentof'Reuenue Noiification s.o.527 aotra i'6tt,1uli, tssi, exceptas respects things doneor omitted to be done .beforesuch supersession,the Cential Gooernmenihereby spectfiesthe qtnntitrl ,nlnt'ionedin coltnnnsS and 6 of the Tablebelow, in relationto.the narcoticdrug or psychotropicsubstnnce mentioned.ii tlr', ,1urrponding entnl in columns2 to 4 of thesaid Table,as the small quantitrl nnd commercialquantity respectiailyfor the purposesof the said clnuse:sof ttrit seciion. TABLE [Seesub-clause vii(a) and xxiii(a) of section 2 of the Act] Sl No. Name of Narcotic Drug and Other non-proprietary Chemical Name Small Commercial Psychotropic Substance name Quanti- Quantity (Intemational non-proprietary ity (in (in gm./kg.) name (INN) Acetorphine 3-0-acetyltetrahydro-7-alpha-(l-hydroxy-l- methylbutyt)-o, l4-endoetheno-onpavine $ 50 9.. 1\) Acetyl-alpha-methylfen N-[-(alpha-methylphenethyl)-4-piperidy]l acetanilide 0.1 g-. J. Acetyldihydrocodeine 100 4. Acetylmethadol 3-acetoxy-6-dimethylamino-4, 4 lheptane 50 gm. 5. Alfentanil -ethyl-4, N-[1-[2-( 5-dihydro-S-oxo-lH-tetrazol-t-yt) 01 gm. ethyll-4-(methoxymethyl)-4-piperidinyll -N- ylpropanamide Allyprodine 3-allyl Jmethyl-4-phenyl-4 Alpha-3-acetoxy-6-dimethylamino-4,Ld lheptane 100 gm Alpha-3-ethyl-l-methyl-4-phenyl-4-propionox 50 gm. 9.7' AlphamethadolArPnametnaool Alpha-6-dimethylamino-4, 4-diphenyl-3-heptanol 2 S0 gm. 10. Alphu-*"thylf"ntur,yl 11. -

Outline for Controlled Substances Program
Environmental Health and Safety Controlled Substances Program Date of Issuance: Review Date: 10/1/2019 (no changes) 10/01/2018 Revision Number: Initial Prepared by: EH&S Table of Contents HEADINGS Introduction Applicability Responsibilities Registration Requirements Authorized Use Ordering/Purchasing Administering and Dispensing Inventory Procedures (Continuing Records) Security Disposal FORMS: Registering or renewing a DEA or state license (CMU) Controlled Substances Authorized users list (CMU) Employee questionnaire for those with access to controlled substances (CMU) Record of Form 222 use (Order form) (CMU) Records of Controlled Substance Purchases (CMU) Record of Controlled Substance Administering and dispensing (CMU) Controlled Substance Physical Inventory (CMU) DEA Registration of Persons doing research or analysis (Form 225) DEA Registration of Dispensers (Form 224) DEA Registration Instructional (Form 224 and 226 to renew) DEA Report of loss or theft (Form 106) DEA Report of drugs surrendered (From 41) DEA SCHEDULES: Schedule I Schedule II Schedule III Schedule IV Schedule V INTRODUCTION State and Federal regulations have been promulgated concerning the use and handling of US Department of Justice Drug Enforcement Administration (DEA) controlled substances. These regulations are in place to address materials which are or have the potential to be addictive or habit forming. These substances have been categorized into “schedules” that have been created by the DEA to reflect their level of concern. The “Carnegie Mellon University DEA Controlled Substances Program” is intended to ensure that Carnegie Mellon University is in compliance with our regulatory requirements. Required activities under the DEA include: 1. Registration of your work with the DEA and with Carnegie Mellon’s Department of Environmental Health and Safety (EH&S). -
![Chapter 329 [New] Uniform Controlled Substances Act](https://docslib.b-cdn.net/cover/1442/chapter-329-new-uniform-controlled-substances-act-1221442.webp)
Chapter 329 [New] Uniform Controlled Substances Act
CHAPTER 329 [NEW] UNIFORM CONTROLLED SUBSTANCES ACT Part I. General Provisions Section 329-1 Definitions 329-2 Hawaii advisory commission on drug abuse and controlled substances; number; appointment 329-3 Annual report 329-4 Duties of the commission Part II. Standards and Schedules 329-11 Authority to schedule controlled substances 329-12 Nomenclature 329-13 Schedule I tests 329-14 Schedule I 329-15 Schedule II tests 329-16 Schedule II 329-17 Schedule III Tests 329-18 Schedule III 329-19 Schedule IV tests 329-20 Schedule IV 329-21 Schedule V tests 329-22 Schedule V 329-23 Republishing and distribution of schedules Part III. Regulation of Manufacture, Distribution, Prescription, and Dispensing of Controlled Substances 329-31 Rules 329-31.5 Clinics 329-32 Registration requirements 329-33 Registration 329-34 Revocation and suspension of registration 329-35 Order to show cause 329-36 Records of registrants 329-37 Filing requirements 329-38 Prescriptions 329-39 Labels 329-40 Methadone treatment programs Part IV. Offenses and Penalties 329-41 Prohibited acts B-penalties 329-42 Prohibited acts C-penalties 329-43 Penalties under other laws 329-43.5 Prohibited acts related to drug paraphernalia Amended 0612 1 329-44 Notice of conviction to be sent to licensing board, department of commerce and consumer affairs 329-45 Repealed 329-46 Prohibited acts related to visits to more than one practitioner to obtain controlled substance prescriptions 329-49 Administrative penalties 329-50 Injunctive relief Part V. Enforcement and Administrative Provisions 329-51 Powers of enforcement personnel 329-52 Administrative inspections 329-53 Injunctions 329-54 Cooperative arrangements and confidentiality 329-55 Forfeitures 329-56 Burden of proof; liabilities 329-57 Judicial review 329-58 Education and research 329-59 Controlled substance registration revolving fund; established Part VI.