Does Cilnidipine, a Dual L- and N-Type Ca2+ Blocker, Shows Promise in Drug Repositioning Approaches?
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Dorset Medicines Advisory Group
DORSET CARDIOLOGY WORKING GROUP GUIDELINE FOR CALCIUM CHANNEL BLOCKERS IN HYPERTENSION SUMMARY The pan-Dorset cardiology working group continues to recommend the use of amlodipine (a third generation dihydropyridine calcium-channel blocker) as first choice calcium channel blocker on the pan-Dorset formulary for hypertension. Lercanidipine is second choice, lacidipine third choice and felodipine is fourth choice. This is due to preferable side effect profiles in terms of ankle oedema and relative costs of the preparations. Note: where angina is the primary indication or is a co-morbidity prescribers must check against the specific product characteristics (SPC) for an individual drug to confirm this is a licensed indication. N.B. Lacidipine and lercandipine are only licensed for use in hypertension. Chapter 02.06.02 CCBs section of the Formulary has undergone an evidence-based review. A comprehensive literature search was carried out on NHS Evidence, Medline, EMBASE, Cochrane Database, and UK Duets. This was for recent reviews or meta-analyses on calcium channel blockers from 2009 onwards (comparative efficacy and side effects) and randomised controlled trials (RCTs). REVIEW BACKGROUND Very little good quality evidence exists. No reviews, meta-analyses or RCTs were found covering all calcium channel blockers currently on the formulary. Another limitation was difficulty obtaining full text original papers for some of the references therefore having to use those from more obscure journals instead. Some discrepancies exist between classification of generations of dihydropyridine CCBs, depending upon the year of publication of the reference/authors’ interpretation. Dihydropyridine (DHP) CCBs tend to be more potent vasodilators than non-dihydropyridine (non-DHP) CCBs (diltiazem, verapamil), but the latter have greater inotropic effects. -
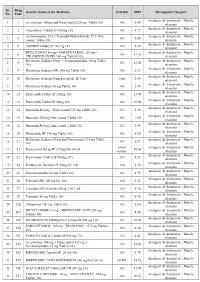
PMBJP Product.Pdf
Sr. Drug Generic Name of the Medicine Unit Size MRP Therapeutic Category No. Code Analgesic & Antipyretic / Muscle 1 1 Aceclofenac 100mg and Paracetamol 325 mg Tablet 10's 10's 8.00 relaxants Analgesic & Antipyretic / Muscle 2 2 Aceclofenac Tablets IP 100mg 10's 10's 4.37 relaxants Acetaminophen 325 + Tramadol Hydrochloride 37.5 film Analgesic & Antipyretic / Muscle 3 4 10's 8.00 coated Tablet 10's relaxants Analgesic & Antipyretic / Muscle 4 5 ASPIRIN Tablets IP 150 mg 14's 14's 2.70 relaxants DICLOFENAC 50 mg+ PARACETAMOL 325 mg+ Analgesic & Antipyretic / Muscle 5 6 10's 11.30 CHLORZOXAZONE 500 mg Tablets 10's relaxants Diclofenac Sodium 50mg + Serratiopeptidase 10mg Tablet Analgesic & Antipyretic / Muscle 6 8 10's 12.00 10's relaxants Analgesic & Antipyretic / Muscle 7 9 Diclofenac Sodium (SR) 100 mg Tablet 10's 10's 6.12 relaxants Analgesic & Antipyretic / Muscle 8 10 Diclofenac Sodium 25mg per ml Inj. IP 3 ml 3 ml 2.00 relaxants Analgesic & Antipyretic / Muscle 9 11 Diclofenac Sodium 50 mg Tablet 10's 10's 2.90 relaxants Analgesic & Antipyretic / Muscle 10 12 Etoricoxilb Tablets IP 120mg 10's 10's 33.00 relaxants Analgesic & Antipyretic / Muscle 11 13 Etoricoxilb Tablets IP 90mg 10's 10's 25.00 relaxants Analgesic & Antipyretic / Muscle 12 14 Ibuprofen 400 mg + Paracetamol 325 mg Tablet 10's 15's 5.50 relaxants Analgesic & Antipyretic / Muscle 13 15 Ibuprofen 200 mg film coated Tablet 10's 10's 1.80 relaxants Analgesic & Antipyretic / Muscle 14 16 Ibuprofen 400 mg film coated Tablet 10's 15's 3.50 relaxants Analgesic & Antipyretic -
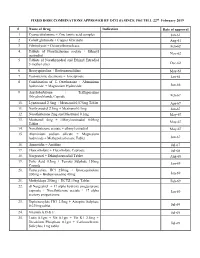
FIXED DOSE COMBINATIONS APPROVED by DCG (I) SINCE 1961 TILL 22Nd February 2019
FIXED DOSE COMBINATIONS APPROVED BY DCG (I) SINCE 1961 TILL 22nd February 2019 # Name of Drug Indication Date of approval 1. Cyanocobalamine + Zinc tannic acid complex Jan-61 2. Cobalt glutamate + Copper Glycinate Aug-61 3. Fibrinolysin + Desoxyribonuclease Feb-62 4. Tablets of Norethisterone acetate + Ethinyl Nov-62 oestradiol 5. Tablets of Norethynodrel and Ethinyl Estradiol 3-methyl ether Dec-62 6. Broxyquinoline + Brobenzoxalidine May-63 7. Testosterone decanoate + Isocaproate Jan-64 8. Combination of L Oxethazaine + Aluminium hydroxide + Magnesium Hydroxide Jun-66 9. Amylobarbitone + Trifluperazine Dihydrochloride Capsule Feb-67 10. Lynestronol 2.5mg + Mestranol 0.075mg Tablet Apr-67 11. Northynodrel 2.5mg + Mestranol 0.1mg Jun-67 12. Norethisterone 2mg and Mestranol 0.1mg May-67 13. Mestranol 4mg + Ethinyloestradial 0.05mg May-67 Tablet 14. Norethisterane acetate + ethinyl estradiol May-67 15. Aluminium sodium silicate + Magnesium hydroxide + Methypolysiloxane Tablet Jun-67 16. Ammoidin + Amidine Jul-67 17. Fluocortolene + Flucortolene Caproate Jul-68 18. Norgestrel + Ethinyloestradiol Tablet Aug-68 19. Folic Acid 0.5mg + Ferrous Sulphate 150mg Jan-69 Capsule 20. Tetracycline HCl 250mg + Broxyquinoline 200mg + Brobenzoxadine 40mg Jan-69 21. Methyldopa 250mg + HCTZ 15mg Tablet Feb-69 22. dl Norgestrel + 17 alpha hydroxy progesterone caproate + Norethisterone acetate + 17 alpha Jan-69 acetoxy progesterone 23. Diphenoxylate HCl 2.5mg + Atropine Sulphate 0.025mg tablet Jul-69 24. Vitamin A,D & E Jul-69 25. Lutin 0.1gm + Vit 0.1gm + Vit K1 2.5mg + Dicalcium Phosphate 0.1gm + Carlozochrome Jul-69 Salicylate 1mg tablet 26. Vit K 1 5mg + Calcium Lactolionate 100 m g+ Carlozocrome Salicylate 2.5mg + Phenol 0.5% Jul-69 + Lignocaine Hcl 1% injection 27. -

Drug Name Plate Number Well Location % Inhibition, Screen Axitinib 1 1 20 Gefitinib (ZD1839) 1 2 70 Sorafenib Tosylate 1 3 21 Cr
Drug Name Plate Number Well Location % Inhibition, Screen Axitinib 1 1 20 Gefitinib (ZD1839) 1 2 70 Sorafenib Tosylate 1 3 21 Crizotinib (PF-02341066) 1 4 55 Docetaxel 1 5 98 Anastrozole 1 6 25 Cladribine 1 7 23 Methotrexate 1 8 -187 Letrozole 1 9 65 Entecavir Hydrate 1 10 48 Roxadustat (FG-4592) 1 11 19 Imatinib Mesylate (STI571) 1 12 0 Sunitinib Malate 1 13 34 Vismodegib (GDC-0449) 1 14 64 Paclitaxel 1 15 89 Aprepitant 1 16 94 Decitabine 1 17 -79 Bendamustine HCl 1 18 19 Temozolomide 1 19 -111 Nepafenac 1 20 24 Nintedanib (BIBF 1120) 1 21 -43 Lapatinib (GW-572016) Ditosylate 1 22 88 Temsirolimus (CCI-779, NSC 683864) 1 23 96 Belinostat (PXD101) 1 24 46 Capecitabine 1 25 19 Bicalutamide 1 26 83 Dutasteride 1 27 68 Epirubicin HCl 1 28 -59 Tamoxifen 1 29 30 Rufinamide 1 30 96 Afatinib (BIBW2992) 1 31 -54 Lenalidomide (CC-5013) 1 32 19 Vorinostat (SAHA, MK0683) 1 33 38 Rucaparib (AG-014699,PF-01367338) phosphate1 34 14 Lenvatinib (E7080) 1 35 80 Fulvestrant 1 36 76 Melatonin 1 37 15 Etoposide 1 38 -69 Vincristine sulfate 1 39 61 Posaconazole 1 40 97 Bortezomib (PS-341) 1 41 71 Panobinostat (LBH589) 1 42 41 Entinostat (MS-275) 1 43 26 Cabozantinib (XL184, BMS-907351) 1 44 79 Valproic acid sodium salt (Sodium valproate) 1 45 7 Raltitrexed 1 46 39 Bisoprolol fumarate 1 47 -23 Raloxifene HCl 1 48 97 Agomelatine 1 49 35 Prasugrel 1 50 -24 Bosutinib (SKI-606) 1 51 85 Nilotinib (AMN-107) 1 52 99 Enzastaurin (LY317615) 1 53 -12 Everolimus (RAD001) 1 54 94 Regorafenib (BAY 73-4506) 1 55 24 Thalidomide 1 56 40 Tivozanib (AV-951) 1 57 86 Fludarabine -

Selection of Effective Cocrystals Former for Dissolution Rate Improvement Of
Selection of effective cocrystals former for dissolution rate improvement of active pharmaceutical ingredients based on lipoaffinity index Piotr Cysewski, Maciej Przybylek To cite this version: Piotr Cysewski, Maciej Przybylek. Selection of effective cocrystals former for dissolution rate im- provement of active pharmaceutical ingredients based on lipoaffinity index. European Journal of Pharmaceutical Sciences, Elsevier, 2017, 107, pp.87-96. 10.1016/j.ejps.2017.07.004. hal-01773096 HAL Id: hal-01773096 https://hal.archives-ouvertes.fr/hal-01773096 Submitted on 29 Apr 2018 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Accepted Manuscript Selection of effective cocrystals former for dissolution rate improvement of active pharmaceutical ingredients based on lipoaffinity index Piotr Cysewski, Maciej Przybyłek PII: S0928-0987(17)30403-7 DOI: doi: 10.1016/j.ejps.2017.07.004 Reference: PHASCI 4127 To appear in: European Journal of Pharmaceutical Sciences Received date: 11 April 2017 Revised date: 6 June 2017 Accepted date: 3 July 2017 Please cite this article as: Piotr Cysewski, Maciej Przybyłek , Selection of effective cocrystals former for dissolution rate improvement of active pharmaceutical ingredients based on lipoaffinity index, European Journal of Pharmaceutical Sciences (2017), doi: 10.1016/j.ejps.2017.07.004 This is a PDF file of an unedited manuscript that has been accepted for publication. -

Cilnidipine, but Not Amlodipine, Ameliorates Osteoporosis in Ovariectomized Hypertensive Rats Through Inhibition of the N-Type Calcium Channel
Hypertension Research (2012) 35, 77–81 & 2012 The Japanese Society of Hypertension All rights reserved 0916-9636/12 www.nature.com/hr ORIGINAL ARTICLE Cilnidipine, but not amlodipine, ameliorates osteoporosis in ovariectomized hypertensive rats through inhibition of the N-type calcium channel Hideo Shimizu1, Hironori Nakagami2, Natsuki Yasumasa2, Osako Kiomy Mariana3, Mariko Kyutoku3, Hiroshi Koriyama3, Futoshi Nakagami3, Munehisa Shimamura2, Hiromi Rakugi1 and Ryuichi Morishita3 Both osteoporosis and high blood pressure are major diseases in aging populations. Recent studies demonstrated that some antihypertensive drugs reduced the risk of bone fracture in elderly patients. Although calcium channel blockers (CCB) are widely used as first-line antihypertensive agents, there is no evidence that they prevent osteoporosis. In this study, we investigated the effects of two types of CCB on bone metabolism: cilnidipine (L-/N-type CCB), which suppresses norepinephrine release from the sympathetic nerve, and amlodipine (L-type CCB). In ovariectomized female spontaneous hypertensive rats, administration of cilnidipine, but not amlodipine, resulted in a significant increase in the ratio of alkaline phosphatase to tartrate-resistant acid phosphatase (TRAP) and a decrease in the number of osteoclasts, as assessed by TRAP staining in the proximal tibia. Bone mineral density, moreover, was significantly higher in the cilnidipine group as compared with the amlodipine group and was associated with a significant decrease in a urinary collagen degradation product (deoxypyridinoline). The degree of prevention of osteoporosis by cilnidipine was similar to that of carvedilol (a b-blocker) because b-blockers reduce fracture risks though the inhibition of osteoclast activation. Interestingly, these effects cannot be attributed to the reduction of blood pressure because all three drugs significantly decreased blood pressure. -

Cardiovascular Calcium Channel Blockers: Historical Overview
Month 2017 Cardiovascular Calcium Channel Blockers: Historical Overview, Development and New Approaches in Design Iryna Drapak,a Lina Perekhoda,b Tetiana Tsapko,b* Natalia Berezniakova,b and Yevgen Tsapkoc aDepartment of General, Bioinorganic, Physical and Colloidal Chemistry, Lvivsˈkyj nacionalˈnyj medychnyj universytet imeni Danyla Halycˈkoho, Lviv 58666, Ukraine bDepartment of Medicinal Chemistry, Nacionalˈnyj farmacevtychnyj universytet, Kharkiv 199318, Ukraine cDepartment of Inorganic Chemistry, Nacionalˈnyj farmacevtychnyj universytet, Kharkiv 199318, Ukraine *E-mail: [email protected]; [email protected] Received July 8, 2016 DOI 10.1002/jhet.2837 Published online 00 Month 2017 in Wiley Online Library (wileyonlinelibrary.com). The data on historical development and modern design approaches for calcium channels blockers (mainly L-type) are summarized in this review. Chemical groups such as dihydropyridine and hydropyrimidine derivatives, as well as phenylalkylamines and benzothiazepines are discussed. J. Heterocyclic Chem., 00, 00 (2017). INTRODUCTION Ca2+. Receptor-operated channels have been defined as those associated with cellular membrane receptors and Role of calcium and calcium channels in vascular smooth activated by specific agonist-receptor interactions. In muscle contraction. Calcium is a key component of the contrast, potential-dependent channels, also known as excitation–contraction coupling process, which occurs voltage-dependent or voltage-gated calcium channels, within the cardiovascular system. It acts as a cellular have been defined as those activated by membrane messenger to link internal or external excitation with depolarization. The Na+/Ca2+ exchange process can cellular response. Increased cytosolic concentrations of promote either influx or efflux because the direction of Ca2+ result in the binding of Ca2+ to a regulatory protein, Ca2+ movement depends upon the relative intracellular either troponin C in cardiac and skeletal muscle or and extracellular ratio of Na+ and Ca2+. -

Serum Triglyceride Lowering Effect of Cilnidipine in Patients with Essential Hypertension
Elmer ress Original Article Cardiol Res. 2016;7(5):173-177 Serum Triglyceride Lowering Effect of Cilnidipine in Patients With Essential Hypertension Prakash Kumara, Arijit Dasb, c, Satish Chandrab, Manju Garib, U. S. P. Keshrib, Kusum Kumarib Abstract the burden is rising owing to escalating obesity and popula- tion aging and by 2025 it is projected that about 1.5 billion of Background: Many epidemiological studies have established the re- patients will be affected worldwide [1]. lationship between hypertension and dyslipidemia. Calcium channel Cardiovascular risk factors, including hypertension, tend blockers (CCBs) are one of the first-line drugs for newly diagnosed to co-segregate more commonly than would be expected by patients with essential hypertension. Cilnidipine as a newer CCB act- chance. Approximately 40% of persons with essential hyper- ing by blocking both L- and N-type calcium channels possesses ad- tension also have dyslipidemia. Genetic studies have estab- ditional beneficial effects apart from lowering blood pressure (BP). lished a clear association between hypertension and dyslipi- The aim of this study was to evaluate the effectiveness of cilnidipine demia [2]. The term dyslipidemic hypertension (DH) was first in patients with essential hypertension with borderline dyslipidemia used in 1988, in the context of familial dyslipidemia along and its effects on lipid profile. with hypertension [3], which was proposed as a genetic syn- drome found in approximately 12% of the patients with es- Methods: Out of 45 enrolled patients, who fulfilled the inclusion cri- sential hypertension and 48% of the hypertensive sib ships [4]. teria, only 37 completed the study. Cilnidipine was started at 10 mg/ Non-familial forms of DH are more common than the famil- day, and then adjusted to 5 - 20 mg/day to achieve the target blood ial ones. -

Stimulatory Effect of Intermittent Hypoxia on the Production Of
www.nature.com/scientificreports OPEN Stimulatory Efect of Intermittent Hypoxia on the Production of Corticosterone by Zona Fasciculata- Received: 23 August 2016 Accepted: 23 June 2017 Reticularis Cells in Rats Published: xx xx xxxx Guey-Shyang Hwang1,9,10, Chih-Chieh Chen1, Jou-Chun Chou2,3, Ling-Ling Chang4, Shu-Fen Kan1, Wei-Ho Lai5, Fu-Kong Lieu5, Sindy Hu8,9,10, Paulus S. Wang1,2,6,7,11 & Shyi-Wu Wang8,10 Hypoxia or intermittent hypoxia (IH) have known to alter both synthesis and secretion of hormones. However, the efect of IH on the production of adrenal cortical steroid hormones is still unclear. The aim of present study was to explore the mechanism involved in the efect of IH on the production of corticosterone by rat ZFR cells. Male rats were exposed at 12% O2 and 88% N2 (8 hours per day) for 1, 2, or 4 days. The ZFR cells were incubated at 37 °C for 1 hour with or without ACTH, 8-Br-cAMP, calcium ion channel blockers, or steroidogenic precursors. The concentration of plasma corticosterone was increased time-dependently by administration of IH hypoxia. The basal levels of corticosterone production in cells were higher in the IH groups than in normoxic group. IH resulted in a time- dependent increase of corticosterone production in response to ACTH, 8-Br-cAMP, progesterone and deoxycorticosterone. The production of pregnenolone in response to 25-OH-C and that of progesterone in response to pregnenolone in ZFR cells were enhanced by 4-day IH. These results suggest that IH in rats increases the secretion of corticosterone via a mechanism at least in part associated with the activation of cAMP pathway and steroidogenic enzymes. -

Cardiovascular Effects of an L/N-Type Ca Channel Blocker Cilnidipine
J Pharmacol Sci 96, 219 – 223 (2004) Journal of Pharmacological Sciences ©2004 The Japanese Pharmacological Society Short Communication Cardiovascular Effects of an L/N-type Ca2+ Channel Blocker Cilnidipine Assessed in the Chronic Atrioventricular Conduction Block Dogs Akira Takahara1,2, Atsushi Sugiyama2,*, Yoshioki Satoh2, Yuji Nakamura2, and Keitaro Hashimoto2 1Pharmaceutical Research Laboratories, Pharmaceuticals Company, Ajinomoto Co., Inc., 1-1 Suzuki-cho, Kawasaki-ku, Kawasaki 210-8681, Japan 2Department of Pharmacology, Interdisciplinary Graduate School of Medicine and Engineering, University of Yamanashi, Tamaho-cho, Nakakoma-gun, Yamanashi 409-3898, Japan Received July 8, 2004; Accepted August 13, 2004 Abstract. Cardiovascular effects of cilnidipine, a dual L/N-type Ca2+ channel blocker, were evaluated in the chronic atrioventricular block dogs, of which systemic blood pressure and plasma catecholamine levels significantly increased in the pre-drug control. Administration of antihypertensive doses of cilnidipine (1 and 3 g/kg, i.v.) significantly decreased the total peri- pheral vascular resistance, mean blood pressure, and atrial rate and increased the cardiac output. These results suggest that cilnidipine not only decreases the blood pressure, but also decreases the sinus automaticity in the in vivo hypertensive condition with increased adrenergic tones. Keywords: cilnidipine, chronic AV block dog, sympathetic nerve activity Cilnidipine is an L/N-type Ca2+ channel blocker used completed within 4 weeks after the onset of AV -

Adverse Drug Reactions Study of Antihypertensive Drugs in Primary Care Settings
JMPF Vol. 10 No. 4 : 241-248 ISSN-p : 2088-8139 ISSN-e : 2443-2946 Adverse Drug Reactions Study of Antihypertensive Drugs in Primary Care Settings Yeni Farida*, Kharimah Faizathus Tsalatsatun Universitas Sebelas Maret, Surakarta, Jawa Tengah Submitted: 17-06-2020 Revised: 24-09-2020 Accepted: 18-12-2020 Korespondensi : Yeni Farida : Email : [email protected] ABSTRACT Hypertension is one of the high-prevalence diseases in primary care. Failure to achieve the target of blood pressure is affected by non-compliance due to the antihypertensive adverse reactions. This study aims to determine adverse drug reaction (ADR) of antihypertensive drugs in primary care settings. A cross sectional study was conducted in “Sibela” Primary Care in Surakarta on March 2019. Investigators interviewed patients directly and observed supporting data from medical records. Hypertension patients with antihypertensive drugs at least for a month were eligible in this study. Then, the data were analyzed by the Liverpool algorithm that interpreted in 4 scales: unlikely, possible, probable, and definite. A total 70 subject were dominated by female (80%). Monotherapy of antihypertensive drugs prescribed to patient in primary care were amlodipine (80%) and captopril (10%). Nine events of ADR were found in hypertension patient. None ADR were doubtful. Possible ADR of amlodipine was drowsiness (5.4%), whereas probable ADR were nausea (3.4%), diuresis (1.8%), and abdominal pain (1.8 %). Definite ADR of captopril was dry mouth (14.3%) and probable ADR was abdominal pain (14.3%). Further investigation regarding the drowsiness, ADR of amlodipine, was needed. Keywords: antihypertensive drugs; Liverpool Algorithm; primary care; ADR INTRODUCTION Study about ADR in hypertension Based on the health profile of Central treatment was important to be conducted for Java province, and especially Surakarta city in optimizing patient safety. -

Stembook 2018.Pdf
The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances FORMER DOCUMENT NUMBER: WHO/PHARM S/NOM 15 WHO/EMP/RHT/TSN/2018.1 © World Health Organization 2018 Some rights reserved. This work is available under the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 IGO licence (CC BY-NC-SA 3.0 IGO; https://creativecommons.org/licenses/by-nc-sa/3.0/igo). Under the terms of this licence, you may copy, redistribute and adapt the work for non-commercial purposes, provided the work is appropriately cited, as indicated below. In any use of this work, there should be no suggestion that WHO endorses any specific organization, products or services. The use of the WHO logo is not permitted. If you adapt the work, then you must license your work under the same or equivalent Creative Commons licence. If you create a translation of this work, you should add the following disclaimer along with the suggested citation: “This translation was not created by the World Health Organization (WHO). WHO is not responsible for the content or accuracy of this translation. The original English edition shall be the binding and authentic edition”. Any mediation relating to disputes arising under the licence shall be conducted in accordance with the mediation rules of the World Intellectual Property Organization. Suggested citation. The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances. Geneva: World Health Organization; 2018 (WHO/EMP/RHT/TSN/2018.1). Licence: CC BY-NC-SA 3.0 IGO. Cataloguing-in-Publication (CIP) data.