Cardiovascular Calcium Channel Blockers: Historical Overview
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

PROZAC Product Monograph Page 1 of 49 Table of Contents
PRODUCT MONOGRAPH PrPROZAC® fluoxetine hydrochloride 10 mg and 20 mg Capsules Antidepressant / Antiobsessional / Antibulimic © Eli Lilly Canada Inc. Date of Revision: January, 25 Exchange Tower 2021 130 King Street West, Suite 900 PO Box 73 Toronto, Ontario M5X 1B1 1-888-545-5972 www.lilly.ca Submission Control No: 192639 PROZAC Product Monograph Page 1 of 49 Table of Contents PART I: HEALTH PROFESSIONAL INFORMATION .......................................................3 SUMMARY PRODUCT INFORMATION...........................................................................3 INDICATIONS AND CLINICAL USE ................................................................................3 CONTRAINDICATIONS .....................................................................................................4 WARNINGS AND PRECAUTIONS ....................................................................................5 ADVERSE REACTIONS ...................................................................................................13 DRUG INTERACTIONS....................................................................................................22 DOSAGE AND ADMINISTRATION ................................................................................27 OVERDOSAGE..................................................................................................................28 ACTION AND CLINICAL PHARMACOLOGY ...............................................................30 STORAGE AND STABILITY............................................................................................32 -
![The Myocardial Metabolic and Haemodynamic Effects of Perhexiline in I]Y Wvo and Nv Vitro Models](https://docslib.b-cdn.net/cover/7158/the-myocardial-metabolic-and-haemodynamic-effects-of-perhexiline-in-i-y-wvo-and-nv-vitro-models-87158.webp)
The Myocardial Metabolic and Haemodynamic Effects of Perhexiline in I]Y Wvo and Nv Vitro Models
THE MYOCARDIAL METABOLIC AND HAEMODYNAMIC EFFECTS OF PERHEXILINE IN I]Y WVO AND NV VITRO MODELS Steven Anthony Unger A Thesis submitted to The University of Adelaide as the requirement for the degree of Doctor of Philosophy The Cardiology Unit, North.Western Adelaide Health Service Department of Medicine, The University of Adelaide. September 2000 u TABLE OF CONTENTS Table of Contents ll Thesis Summary vlt Dcclaration ix Acknowledgments x CHAPTER 1: A LITERATURE REVIEW OF METABOLIC APPROACHES TO MYOCARDIAL ISCHAEMIA I 1.1 INTRODUCTION 2 7.2 CHRONIC MYOCARDIAL ISCHAEMIA 3 1.2.1 Epidemiology 3 1.2.2 Pathogenesis 4 1.3 CARDIAC METABOLISM 7 1.3.1 Overview offatty acid metabolism 8 1.3.1.1 Uptake and transport within the aqueous cytoplasm 8 1.3.1.2 Transport into mitochondria 9 1.3.1.3 B-oxidation l0 1.3.1.4 TCA cycle and electron transport chain 10 1.3.2 Regulation offatty acid metabolism 11 1.3.3 Oventiew of carbohydrate metabolism 13 1.3.4 Regulation of carbohydrate metabolism 14 ' L3.5 Substrate utilisation by the heart: the cost,in oxygen 16 1.3.6 Myocardial metabolism during ischaemia/reperfusion 18 1.3.7 The role offatty acid metabolites 22 1.4 MANAGEMENT OF CHRONIC MYOCARDIAL ISCHAEMIA 24 1.4.1 Medical therapy 24 alaaìf:a--.--nÁ 1.4. i. i i\iüa[es L+ 1.4.1.2 B-adrenoceptor antagonists 25 lrl 1.4.1.3 L-t1pe calcium antagonists 25 1.4.2 Revascularisation 26 1.4.2.1 Percutaneous transluminal coronary angioplasty 26 1.4.2.2 Coronary artery b¡pass surgery 28 I .4.2.3 Transmyocardial laser revascularisation 29 1.4.3 Limitations of current strategies 30 1.5 METABOLIC APPROACHES TO MYOCARDIAL ISCHAEMIA 32 1.5.1 Increasing glucose supply to the heart 35 1.5. -

2D6 Substrates 2D6 Inhibitors 2D6 Inducers
Physician Guidelines: Drugs Metabolized by Cytochrome P450’s 1 2D6 Substrates Acetaminophen Captopril Dextroamphetamine Fluphenazine Methoxyphenamine Paroxetine Tacrine Ajmaline Carteolol Dextromethorphan Fluvoxamine Metoclopramide Perhexiline Tamoxifen Alprenolol Carvedilol Diazinon Galantamine Metoprolol Perphenazine Tamsulosin Amiflamine Cevimeline Dihydrocodeine Guanoxan Mexiletine Phenacetin Thioridazine Amitriptyline Chloropromazine Diltiazem Haloperidol Mianserin Phenformin Timolol Amphetamine Chlorpheniramine Diprafenone Hydrocodone Minaprine Procainamide Tolterodine Amprenavir Chlorpyrifos Dolasetron Ibogaine Mirtazapine Promethazine Tradodone Aprindine Cinnarizine Donepezil Iloperidone Nefazodone Propafenone Tramadol Aripiprazole Citalopram Doxepin Imipramine Nifedipine Propranolol Trimipramine Atomoxetine Clomipramine Encainide Indoramin Nisoldipine Quanoxan Tropisetron Benztropine Clozapine Ethylmorphine Lidocaine Norcodeine Quetiapine Venlafaxine Bisoprolol Codeine Ezlopitant Loratidine Nortriptyline Ranitidine Verapamil Brofaramine Debrisoquine Flecainide Maprotline olanzapine Remoxipride Zotepine Bufuralol Delavirdine Flunarizine Mequitazine Ondansetron Risperidone Zuclopenthixol Bunitrolol Desipramine Fluoxetine Methadone Oxycodone Sertraline Butylamphetamine Dexfenfluramine Fluperlapine Methamphetamine Parathion Sparteine 2D6 Inhibitors Ajmaline Chlorpromazine Diphenhydramine Indinavir Mibefradil Pimozide Terfenadine Amiodarone Cimetidine Doxorubicin Lasoprazole Moclobemide Quinidine Thioridazine Amitriptyline Cisapride -

Dorset Medicines Advisory Group
DORSET CARDIOLOGY WORKING GROUP GUIDELINE FOR CALCIUM CHANNEL BLOCKERS IN HYPERTENSION SUMMARY The pan-Dorset cardiology working group continues to recommend the use of amlodipine (a third generation dihydropyridine calcium-channel blocker) as first choice calcium channel blocker on the pan-Dorset formulary for hypertension. Lercanidipine is second choice, lacidipine third choice and felodipine is fourth choice. This is due to preferable side effect profiles in terms of ankle oedema and relative costs of the preparations. Note: where angina is the primary indication or is a co-morbidity prescribers must check against the specific product characteristics (SPC) for an individual drug to confirm this is a licensed indication. N.B. Lacidipine and lercandipine are only licensed for use in hypertension. Chapter 02.06.02 CCBs section of the Formulary has undergone an evidence-based review. A comprehensive literature search was carried out on NHS Evidence, Medline, EMBASE, Cochrane Database, and UK Duets. This was for recent reviews or meta-analyses on calcium channel blockers from 2009 onwards (comparative efficacy and side effects) and randomised controlled trials (RCTs). REVIEW BACKGROUND Very little good quality evidence exists. No reviews, meta-analyses or RCTs were found covering all calcium channel blockers currently on the formulary. Another limitation was difficulty obtaining full text original papers for some of the references therefore having to use those from more obscure journals instead. Some discrepancies exist between classification of generations of dihydropyridine CCBs, depending upon the year of publication of the reference/authors’ interpretation. Dihydropyridine (DHP) CCBs tend to be more potent vasodilators than non-dihydropyridine (non-DHP) CCBs (diltiazem, verapamil), but the latter have greater inotropic effects. -

Drug-Induced Fatty Liver Disease
789 Drug-induced fatty liver disease. , 2015; 14CONCISE (6): 789-806 REVIEW November-December, Vol. 14 No. 6, 2015: 789-806 Drug-induced fatty liver disease: An overview of pathogenesis and management Sanjaya K. Satapathy,* Vanessa Kuwajima,** Jeffrey Nadelson,** Omair Atiq,*** Arun J. Sanyal**** * Methodist University Hospital Transplant Institute, Division of Surgery, University of Tennessee Health Sciences Center, Memphis, Tennessee, USA. ** Division of Gastroenterology and Hepatology, University of Tennessee Health Sciences Center, Memphis, Tennessee, USA. *** University of Texas Southwestern, Dallas, Texas, USA. **** Division of Gastroenterology, Hepatology and Nutrition, Virginia Commonwealth University Health System, Richmond, Virginia, USA. ABSTRACT Over the past decades, many drugs have been identified, that can potentially induce steatohepatitis in the predisposed individual. Classically this has been incriminated to amiodarone, perhexiline, and 4,4’-diethyla- minoethoxyhexestrol (DH), all of which have been found to independently induce the histologic picture of non-alcoholic steatohepatitis (NASH). Pathogenetic mechanisms of hepatotoxicity although still evolving, demonstrate that mitochondrial dysfunction, deranged ATP production and fatty acid catabolism likely play an important role. Drugs like steroid hormones can exacerbate the pathogenetic mechanisms that lead to NASH, and other drugs like tamoxifen, cisplatin and irenotecan have been shown to precipitate la- tent fatty liver as well. Further research aiming to elucidate -
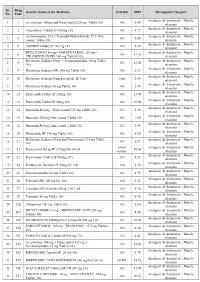
PMBJP Product.Pdf
Sr. Drug Generic Name of the Medicine Unit Size MRP Therapeutic Category No. Code Analgesic & Antipyretic / Muscle 1 1 Aceclofenac 100mg and Paracetamol 325 mg Tablet 10's 10's 8.00 relaxants Analgesic & Antipyretic / Muscle 2 2 Aceclofenac Tablets IP 100mg 10's 10's 4.37 relaxants Acetaminophen 325 + Tramadol Hydrochloride 37.5 film Analgesic & Antipyretic / Muscle 3 4 10's 8.00 coated Tablet 10's relaxants Analgesic & Antipyretic / Muscle 4 5 ASPIRIN Tablets IP 150 mg 14's 14's 2.70 relaxants DICLOFENAC 50 mg+ PARACETAMOL 325 mg+ Analgesic & Antipyretic / Muscle 5 6 10's 11.30 CHLORZOXAZONE 500 mg Tablets 10's relaxants Diclofenac Sodium 50mg + Serratiopeptidase 10mg Tablet Analgesic & Antipyretic / Muscle 6 8 10's 12.00 10's relaxants Analgesic & Antipyretic / Muscle 7 9 Diclofenac Sodium (SR) 100 mg Tablet 10's 10's 6.12 relaxants Analgesic & Antipyretic / Muscle 8 10 Diclofenac Sodium 25mg per ml Inj. IP 3 ml 3 ml 2.00 relaxants Analgesic & Antipyretic / Muscle 9 11 Diclofenac Sodium 50 mg Tablet 10's 10's 2.90 relaxants Analgesic & Antipyretic / Muscle 10 12 Etoricoxilb Tablets IP 120mg 10's 10's 33.00 relaxants Analgesic & Antipyretic / Muscle 11 13 Etoricoxilb Tablets IP 90mg 10's 10's 25.00 relaxants Analgesic & Antipyretic / Muscle 12 14 Ibuprofen 400 mg + Paracetamol 325 mg Tablet 10's 15's 5.50 relaxants Analgesic & Antipyretic / Muscle 13 15 Ibuprofen 200 mg film coated Tablet 10's 10's 1.80 relaxants Analgesic & Antipyretic / Muscle 14 16 Ibuprofen 400 mg film coated Tablet 10's 15's 3.50 relaxants Analgesic & Antipyretic -

Drug-Induced Inhibition of Mitochondrial Fatty Acid Oxidation
Drug-Induced Inhibition of Mitochondrial Fatty Acid Oxidation and Steatosis Julie Massart, Karima Begriche, Nelly Buron, Mathieu Porceddu, Annie Borgne-Sanchez, Bernard Fromenty To cite this version: Julie Massart, Karima Begriche, Nelly Buron, Mathieu Porceddu, Annie Borgne-Sanchez, et al.. Drug- Induced Inhibition of Mitochondrial Fatty Acid Oxidation and Steatosis. Current Pathobiology Re- ports, Springer, 2013, 1 (3), pp.147-157. 10.1007/s40139-013-0022-y. hal-00860237 HAL Id: hal-00860237 https://hal-univ-rennes1.archives-ouvertes.fr/hal-00860237 Submitted on 10 Sep 2013 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Heading for SpringerLink.com: Mitochondrial Dysfunction and Diseases (H Jaeschke, Section Editor) Drug-Induced Inhibition of Mitochondrial Fatty Acid Oxidation and Steatosis Julie Massart Karima Begriche Nelly Buron Mathieu Porceddu Annie Borgne-Sanchez Bernard Fromenty K. Begriche B. Fromenty () INSERM, U991, Université de Rennes 1 2, avenue du Professeur Léon Bernard 35043 Rennes Cedex, France e-mail: [email protected] -

Abnormal Laboratory Results Therapeutic Drug Monitoring
Abnormal laboratory results Therapeutic drug monitoring: which drugs, why, when and how to do it RA Ghiculescu, Senior Clinical Pharmacology Registrar, Department of Clinical Pharmacology, Princess Alexandra Hospital, Brisbane Summary Which drugs? Therapeutic drug monitoring of concentrations of When an effect, such as changes in blood pressure, pain or drugs in body fluids, usually plasma, can be used serum cholesterol is readily measured, the dose of a drug during treatment and for diagnostic purposes. The should be adjusted according to the response. Monitoring drug concentration is more useful when drugs are used to selection of drugs for therapeutic drug monitoring prevent an adverse outcome, for example, graft rejection or to is important as the concentrations of many avoid toxicity, as with aminoglycosides. A drug should satisfy drugs are not clearly related to their effects. For certain criteria to be suitable for therapeutic drug monitoring. selected drugs therapeutic drug monitoring aims Examples include: to enhance drug efficacy, reduce toxicity or assist n narrow target range with diagnosis. Despite its apparent advantages, n significant pharmacokinetic variability it has inherent limitations. Some large hospitals n a reasonable relationship between plasma concentrations have services which provide support with drug and clinical effects monitoring and interpretation of results. n established target concentration range Key words: pharmacokinetics. n availability of cost-effective drug assay. (Aust Prescr 2008;31:42–4) The most commonly monitored drugs are probably Introduction carbamazepine, valproate and digoxin. However, there is little The monitoring of therapeutic drugs involves measuring drug evidence that monitoring concentrations of anticonvulsants concentrations in plasma, serum or blood. -

DOMASYS Standard
New Zealand Data Sheet Optisulin – insulin glargine NEW ZEALAND DATA SHEET 1 PRODUCT NAME Optisulin 100 IU/mL solution for injection in 10 mL vials. Optisulin 100 IU/mL solution for injection in 3 mL cartridges. Optisulin SoloStar 100 IU/mL solution for injection in a 3 mL pre-filled pen. 2 QUALITATIVE AND QUANTITATIVE COMPOSITION Optisulin contains 100 IU/mL (3.6378 mg/mL) insulin glargine. Optisulin 100 IU/mL solution for injection in 10 mL vials - equivalent to 1000 IU. Optisulin 100 IU/mL solution for injection in 3 mL cartridges - equivalent to 300 IU. Optisulin SoloStar 100 IU/mL solution for injection in a 3 mL pre-filled pen – equivalent to 300 IU. Insulin glargine is produced by recombinant DNA technology in Escherichia coli. For the full list of excipients, see section 6.1. 3 PHARMACEUTICAL FORM Optisulin is a sterile solution of insulin glargine in vials and cartridges for use as an injection. 4 CLINICAL PARTICULARS 4.1 THERAPEUTIC INDICATIONS Optisulin is an insulin analogue indicated for once-daily subcutaneous administration in the treatment of type 1 or type 2 diabetes mellitus patients who require insulin for the control of optisulin-ccdsv20-dsv2-27jan21 Page 1 New Zealand Data Sheet Optisulin – insulin glargine hyperglycaemia. 4.2 DOSE AND METHOD OF ADMINISTRATION Dose Optisulin is an insulin analogue, equipotent to human insulin, with a peakless glucose lowering profile and a prolonged duration of action that permits once daily dosing. The desired blood glucose levels as well as the doses and timing of any antidiabetic medication, including Optisulin, must be determined and adjusted individually. -
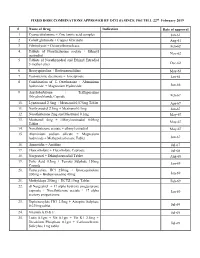
FIXED DOSE COMBINATIONS APPROVED by DCG (I) SINCE 1961 TILL 22Nd February 2019
FIXED DOSE COMBINATIONS APPROVED BY DCG (I) SINCE 1961 TILL 22nd February 2019 # Name of Drug Indication Date of approval 1. Cyanocobalamine + Zinc tannic acid complex Jan-61 2. Cobalt glutamate + Copper Glycinate Aug-61 3. Fibrinolysin + Desoxyribonuclease Feb-62 4. Tablets of Norethisterone acetate + Ethinyl Nov-62 oestradiol 5. Tablets of Norethynodrel and Ethinyl Estradiol 3-methyl ether Dec-62 6. Broxyquinoline + Brobenzoxalidine May-63 7. Testosterone decanoate + Isocaproate Jan-64 8. Combination of L Oxethazaine + Aluminium hydroxide + Magnesium Hydroxide Jun-66 9. Amylobarbitone + Trifluperazine Dihydrochloride Capsule Feb-67 10. Lynestronol 2.5mg + Mestranol 0.075mg Tablet Apr-67 11. Northynodrel 2.5mg + Mestranol 0.1mg Jun-67 12. Norethisterone 2mg and Mestranol 0.1mg May-67 13. Mestranol 4mg + Ethinyloestradial 0.05mg May-67 Tablet 14. Norethisterane acetate + ethinyl estradiol May-67 15. Aluminium sodium silicate + Magnesium hydroxide + Methypolysiloxane Tablet Jun-67 16. Ammoidin + Amidine Jul-67 17. Fluocortolene + Flucortolene Caproate Jul-68 18. Norgestrel + Ethinyloestradiol Tablet Aug-68 19. Folic Acid 0.5mg + Ferrous Sulphate 150mg Jan-69 Capsule 20. Tetracycline HCl 250mg + Broxyquinoline 200mg + Brobenzoxadine 40mg Jan-69 21. Methyldopa 250mg + HCTZ 15mg Tablet Feb-69 22. dl Norgestrel + 17 alpha hydroxy progesterone caproate + Norethisterone acetate + 17 alpha Jan-69 acetoxy progesterone 23. Diphenoxylate HCl 2.5mg + Atropine Sulphate 0.025mg tablet Jul-69 24. Vitamin A,D & E Jul-69 25. Lutin 0.1gm + Vit 0.1gm + Vit K1 2.5mg + Dicalcium Phosphate 0.1gm + Carlozochrome Jul-69 Salicylate 1mg tablet 26. Vit K 1 5mg + Calcium Lactolionate 100 m g+ Carlozocrome Salicylate 2.5mg + Phenol 0.5% Jul-69 + Lignocaine Hcl 1% injection 27. -

Drug Name Plate Number Well Location % Inhibition, Screen Axitinib 1 1 20 Gefitinib (ZD1839) 1 2 70 Sorafenib Tosylate 1 3 21 Cr
Drug Name Plate Number Well Location % Inhibition, Screen Axitinib 1 1 20 Gefitinib (ZD1839) 1 2 70 Sorafenib Tosylate 1 3 21 Crizotinib (PF-02341066) 1 4 55 Docetaxel 1 5 98 Anastrozole 1 6 25 Cladribine 1 7 23 Methotrexate 1 8 -187 Letrozole 1 9 65 Entecavir Hydrate 1 10 48 Roxadustat (FG-4592) 1 11 19 Imatinib Mesylate (STI571) 1 12 0 Sunitinib Malate 1 13 34 Vismodegib (GDC-0449) 1 14 64 Paclitaxel 1 15 89 Aprepitant 1 16 94 Decitabine 1 17 -79 Bendamustine HCl 1 18 19 Temozolomide 1 19 -111 Nepafenac 1 20 24 Nintedanib (BIBF 1120) 1 21 -43 Lapatinib (GW-572016) Ditosylate 1 22 88 Temsirolimus (CCI-779, NSC 683864) 1 23 96 Belinostat (PXD101) 1 24 46 Capecitabine 1 25 19 Bicalutamide 1 26 83 Dutasteride 1 27 68 Epirubicin HCl 1 28 -59 Tamoxifen 1 29 30 Rufinamide 1 30 96 Afatinib (BIBW2992) 1 31 -54 Lenalidomide (CC-5013) 1 32 19 Vorinostat (SAHA, MK0683) 1 33 38 Rucaparib (AG-014699,PF-01367338) phosphate1 34 14 Lenvatinib (E7080) 1 35 80 Fulvestrant 1 36 76 Melatonin 1 37 15 Etoposide 1 38 -69 Vincristine sulfate 1 39 61 Posaconazole 1 40 97 Bortezomib (PS-341) 1 41 71 Panobinostat (LBH589) 1 42 41 Entinostat (MS-275) 1 43 26 Cabozantinib (XL184, BMS-907351) 1 44 79 Valproic acid sodium salt (Sodium valproate) 1 45 7 Raltitrexed 1 46 39 Bisoprolol fumarate 1 47 -23 Raloxifene HCl 1 48 97 Agomelatine 1 49 35 Prasugrel 1 50 -24 Bosutinib (SKI-606) 1 51 85 Nilotinib (AMN-107) 1 52 99 Enzastaurin (LY317615) 1 53 -12 Everolimus (RAD001) 1 54 94 Regorafenib (BAY 73-4506) 1 55 24 Thalidomide 1 56 40 Tivozanib (AV-951) 1 57 86 Fludarabine -

Repositioning Fda-Approved Drugs in Combination with Epigenetic Drugs to Reprogram Colon Cancer Epigenome
Author Manuscript Published OnlineFirst on December 15, 2016; DOI: 10.1158/1535-7163.MCT-16-0588 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. REPOSITIONING FDA-APPROVED DRUGS IN COMBINATION WITH EPIGENETIC DRUGS TO REPROGRAM COLON CANCER EPIGENOME Noël J.-M. Raynal1,2, Elodie M. Da Costa2, Justin T. Lee1, Vazganush Gharibyan3, Saira Ahmed3, Hanghang Zhang1,Takahiro Sato1, Gabriel G. Malouf4, and Jean-Pierre J. Issa1 1Fels Institute for Cancer Research and Molecular Biology, Temple University School of Medicine, 3307 North Broad Street, Philadelphia, PA, 19140, USA. 2Département de pharmacologie, Université de Montréal and Sainte-Justine University Hospital Research Center, 3175, Chemin de la Côte-Sainte- Catherine, Montréal (Québec) H3T 1C5, Canada. 3Department of Leukemia, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Blvd., Houston, TX, 77030, USA. 4Department of Medical Oncology, Groupe Hospitalier Pitié-Salpêtrière, University Pierre and Marie Curie (Paris VI), Institut Universitaire de cancérologie, AP-HP, Paris, France. Note: Supplementary data for this article are available at Molecular Cancer Therapeutics Online (http://mct.aacrjournals.org/). Corresponding Author: Noël J.-M. Raynal, Département de Pharmacologie, Université de Montréal , Centre de recherche de l’Hôpital Sainte-Justine, 3175, Chemin de la Côte-Sainte-Catherine, Montréal (Québec), H3T 1C5, Canada. Phone : (514) 345-4931 ext. 6763. Email : [email protected] Running title: High-throughput screening for epigenetic drug combinations Key words: Drug repurposing, Drug combination, High-throughput drug screening, Epigenetic therapy The authors declare no potential conflicts of interest. 1 Downloaded from mct.aacrjournals.org on September 23, 2021.