Identifying Genomic Biomarkers for Cancer Treatment
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Cellular and Molecular Signatures in the Disease Tissue of Early
Cellular and Molecular Signatures in the Disease Tissue of Early Rheumatoid Arthritis Stratify Clinical Response to csDMARD-Therapy and Predict Radiographic Progression Frances Humby1,* Myles Lewis1,* Nandhini Ramamoorthi2, Jason Hackney3, Michael Barnes1, Michele Bombardieri1, Francesca Setiadi2, Stephen Kelly1, Fabiola Bene1, Maria di Cicco1, Sudeh Riahi1, Vidalba Rocher-Ros1, Nora Ng1, Ilias Lazorou1, Rebecca E. Hands1, Desiree van der Heijde4, Robert Landewé5, Annette van der Helm-van Mil4, Alberto Cauli6, Iain B. McInnes7, Christopher D. Buckley8, Ernest Choy9, Peter Taylor10, Michael J. Townsend2 & Costantino Pitzalis1 1Centre for Experimental Medicine and Rheumatology, William Harvey Research Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, UK. Departments of 2Biomarker Discovery OMNI, 3Bioinformatics and Computational Biology, Genentech Research and Early Development, South San Francisco, California 94080 USA 4Department of Rheumatology, Leiden University Medical Center, The Netherlands 5Department of Clinical Immunology & Rheumatology, Amsterdam Rheumatology & Immunology Center, Amsterdam, The Netherlands 6Rheumatology Unit, Department of Medical Sciences, Policlinico of the University of Cagliari, Cagliari, Italy 7Institute of Infection, Immunity and Inflammation, University of Glasgow, Glasgow G12 8TA, UK 8Rheumatology Research Group, Institute of Inflammation and Ageing (IIA), University of Birmingham, Birmingham B15 2WB, UK 9Institute of -

WO 2019/068007 Al Figure 2
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization I International Bureau (10) International Publication Number (43) International Publication Date WO 2019/068007 Al 04 April 2019 (04.04.2019) W 1P O PCT (51) International Patent Classification: (72) Inventors; and C12N 15/10 (2006.01) C07K 16/28 (2006.01) (71) Applicants: GROSS, Gideon [EVIL]; IE-1-5 Address C12N 5/10 (2006.0 1) C12Q 1/6809 (20 18.0 1) M.P. Korazim, 1292200 Moshav Almagor (IL). GIBSON, C07K 14/705 (2006.01) A61P 35/00 (2006.01) Will [US/US]; c/o ImmPACT-Bio Ltd., 2 Ilian Ramon St., C07K 14/725 (2006.01) P.O. Box 4044, 7403635 Ness Ziona (TL). DAHARY, Dvir [EilL]; c/o ImmPACT-Bio Ltd., 2 Ilian Ramon St., P.O. (21) International Application Number: Box 4044, 7403635 Ness Ziona (IL). BEIMAN, Merav PCT/US2018/053583 [EilL]; c/o ImmPACT-Bio Ltd., 2 Ilian Ramon St., P.O. (22) International Filing Date: Box 4044, 7403635 Ness Ziona (E.). 28 September 2018 (28.09.2018) (74) Agent: MACDOUGALL, Christina, A. et al; Morgan, (25) Filing Language: English Lewis & Bockius LLP, One Market, Spear Tower, SanFran- cisco, CA 94105 (US). (26) Publication Language: English (81) Designated States (unless otherwise indicated, for every (30) Priority Data: kind of national protection available): AE, AG, AL, AM, 62/564,454 28 September 2017 (28.09.2017) US AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, 62/649,429 28 March 2018 (28.03.2018) US CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, (71) Applicant: IMMP ACT-BIO LTD. -

Pbmcs Transcriptome Profiles Identified Breed-Specific Transcriptome Signatures for PRRSV Vaccination in German Landrace and Pietrain Pigs
RESEARCH ARTICLE PBMCs transcriptome profiles identified breed-specific transcriptome signatures for PRRSV vaccination in German Landrace and Pietrain pigs 1,2 1 1,3 Md. Aminul IslamID , Christiane Neuhoff , Sharmin Aqter RonyID , Christine Groûe- Brinkhaus1, Muhammad Jasim Uddin2,4, Michael HoÈ lker1,5, Dawit Tesfaye1, Ernst Tholen1, 1,5 1 Karl Schellander , Maren Julia ProÈ ll-CornelissenID * a1111111111 a1111111111 1 Department of Animal Breeding and Husbandry, Institute of Animal Science, University of Bonn, Endenicher Allee 15, Bonn, Germany, 2 Department of Medicine, Faculty of Veterinary Science, Bangladesh a1111111111 Agricultural University, Mymensingh, Bangladesh, 3 Department of Parasitology, Faculty of Veterinary a1111111111 Science, Bangladesh Agricultural University, Mymensingh, Bangladesh, 4 School of Veterinary Science, The a1111111111 University of Queensland, Gatton campus, Brisbane, QLD, Australia, 5 Teaching and Research Station on Frankenforst, Faculty of Agriculture, University of Bonn, KoÈnigswinter, Germany * [email protected] OPEN ACCESS Citation: Islam M.A, Neuhoff C, Aqter Rony S, Abstract Groûe-Brinkhaus C, Uddin MJ, HoÈlker M, et al. (2019) PBMCs transcriptome profiles identified Porcine reproductive and respiratory syndrome (PRRS) is a devastating viral disease affect- breed-specific transcriptome signatures for PRRSV ing the swine industry worldwide. Genetic variation in host immunity has been considered vaccination in German Landrace and Pietrain pigs. as one of the potential determinants to improve the immunocompetence, thereby resistance PLoS ONE 14(9): e0222513. https://doi.org/ to PRRS. Therefore, the present study aimed to investigate the breed difference in innate 10.1371/journal.pone.0222513 immune response to PRRSV vaccination between German Landrace (DL) and Pietrain (Pi) Editor: Juan J. Loor, University of Illinois, UNITED pigs. -

Us 2018 / 0305689 A1
US 20180305689A1 ( 19 ) United States (12 ) Patent Application Publication ( 10) Pub . No. : US 2018 /0305689 A1 Sætrom et al. ( 43 ) Pub . Date: Oct. 25 , 2018 ( 54 ) SARNA COMPOSITIONS AND METHODS OF plication No . 62 /150 , 895 , filed on Apr. 22 , 2015 , USE provisional application No . 62/ 150 ,904 , filed on Apr. 22 , 2015 , provisional application No. 62 / 150 , 908 , (71 ) Applicant: MINA THERAPEUTICS LIMITED , filed on Apr. 22 , 2015 , provisional application No. LONDON (GB ) 62 / 150 , 900 , filed on Apr. 22 , 2015 . (72 ) Inventors : Pål Sætrom , Trondheim (NO ) ; Endre Publication Classification Bakken Stovner , Trondheim (NO ) (51 ) Int . CI. C12N 15 / 113 (2006 .01 ) (21 ) Appl. No. : 15 /568 , 046 (52 ) U . S . CI. (22 ) PCT Filed : Apr. 21 , 2016 CPC .. .. .. C12N 15 / 113 ( 2013 .01 ) ; C12N 2310 / 34 ( 2013. 01 ) ; C12N 2310 /14 (2013 . 01 ) ; C12N ( 86 ) PCT No .: PCT/ GB2016 /051116 2310 / 11 (2013 .01 ) $ 371 ( c ) ( 1 ) , ( 2 ) Date : Oct . 20 , 2017 (57 ) ABSTRACT The invention relates to oligonucleotides , e . g . , saRNAS Related U . S . Application Data useful in upregulating the expression of a target gene and (60 ) Provisional application No . 62 / 150 ,892 , filed on Apr. therapeutic compositions comprising such oligonucleotides . 22 , 2015 , provisional application No . 62 / 150 ,893 , Methods of using the oligonucleotides and the therapeutic filed on Apr. 22 , 2015 , provisional application No . compositions are also provided . 62 / 150 ,897 , filed on Apr. 22 , 2015 , provisional ap Specification includes a Sequence Listing . SARNA sense strand (Fessenger 3 ' SARNA antisense strand (Guide ) Mathew, Si Target antisense RNA transcript, e . g . NAT Target Coding strand Gene Transcription start site ( T55 ) TY{ { ? ? Targeted Target transcript , e . -

WO 2016/004387 Al 7 January 2016 (07.01.2016) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/004387 Al 7 January 2016 (07.01.2016) P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every A61P 35/00 (2006.01) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, (21) International Application Number: BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, PCT/US20 15/039 108 DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (22) International Filing Date: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, 2 July 2015 (02.07.2015) KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (25) Filing Language: English PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, (26) Publication Language: English SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: 62/020,3 10 2 July 2014 (02.07.2014) US (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (71) Applicant: H. LEE MOFFITT CANCER CENTER GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, AND RESEARCH INSTITUTE, INC. [US/US]; 12902 TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, Magnolia Dr., Tampa, FL 336 12-9497 (US). -

Supplementary Tables
Supplementary Tables Supplementary Table S1: Clinicopathologic characteristics of 64 patients with untreated primary myxofibrosarcoma Characteristic n (%) or median (range) Gender Male 23 (36%) Female 41 (64%) Age, years 63.5 (24-88) Location Extremity 55 (86%) Trunk/chest wall 8 (13%) Retroperitoneal 1 (1%) Size, cm ≤5 9 (14%) >5; ≤10 34 (53%) >10 21 (33%) Median 8.5 (3.1-25) Depth Superficial 12 (19%) Deep 52 (81%) Surgical margins R0 49 (77%) R1 15 (23%) Percent Myxoid 75% (10%-100%) Local Recurrence 14 (22%) Distant Recurrence 22 (34%) Died of Disease 17 (27%) Follow-up, years All patients 2.6 (0.1-9.2) Surviving patients 3.1 (0.4-9.2) Time to death, years 1.8 (0.7-3.3) 213338 at RIS1 3.00E-06 -2.45 219840 s at TCL6 1.30E-05 -0.25 201645 at TNC 2.00E-06 -2.15 208052 x at CEACAM3 1.40E-05 -0.25 205542 at STEAP1 1.70E-05 -1.74 210339 s at KLK2 1.40E-05 -0.25 212473 s at MICAL2 2.20E-05 -1.6 206612 at CACNG1 1.50E-05 -0.25 204337 at RGS4 <1.00E-06 -1.51 205531 s at GLS2 1.60E-05 -0.25 208025 s at HMGA2 4.50E-05 -1.51 215274 at SLC12A3 1.60E-05 -0.25 204464 s at EDNRA 6.00E-05 -1.48 216881 x at PRB4 1.60E-05 -0.25 208712 at CCND1 1.20E-05 -1.47 206869 at CHAD 1.80E-05 -0.25 201506 at TGFBI <1.00E-06 -1.32 207528 s at SLC7A11 1.80E-05 -0.25 203324 s at CAV2 <1.00E-06 -1.29 210577 at CASR 1.80E-05 -0.25 201617 x at CALD1 5.00E-06 -1.24 205815 at REG3A 1.90E-05 -0.25 206766 at ITGA10 2.60E-05 -1.23 208204 s at CAV3 1.90E-05 -0.25 209218 at SQLE <1.00E-06 -1.18 220581 at C6orf97 2.00E-05 -0.25 221773 at ELK3 <1.00E-06 -1.14 207007 at NR1I3 -
Explorations in Olfactory Receptor Structure and Function by Jianghai
Explorations in Olfactory Receptor Structure and Function by Jianghai Ho Department of Neurobiology Duke University Date:_______________________ Approved: ___________________________ Hiroaki Matsunami, Supervisor ___________________________ Jorg Grandl, Chair ___________________________ Marc Caron ___________________________ Sid Simon ___________________________ [Committee Member Name] Dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Neurobiology in the Graduate School of Duke University 2014 ABSTRACT Explorations in Olfactory Receptor Structure and Function by Jianghai Ho Department of Neurobiology Duke University Date:_______________________ Approved: ___________________________ Hiroaki Matsunami, Supervisor ___________________________ Jorg Grandl, Chair ___________________________ Marc Caron ___________________________ Sid Simon ___________________________ [Committee Member Name] An abstract of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Neurobiology in the Graduate School of Duke University 2014 Copyright by Jianghai Ho 2014 Abstract Olfaction is one of the most primitive of our senses, and the olfactory receptors that mediate this very important chemical sense comprise the largest family of genes in the mammalian genome. It is therefore surprising that we understand so little of how olfactory receptors work. In particular we have a poor idea of what chemicals are detected by most of the olfactory receptors in the genome, and for those receptors which we have paired with ligands, we know relatively little about how the structure of these ligands can either activate or inhibit the activation of these receptors. Furthermore the large repertoire of olfactory receptors, which belong to the G protein coupled receptor (GPCR) superfamily, can serve as a model to contribute to our broader understanding of GPCR-ligand binding, especially since GPCRs are important pharmaceutical targets. -
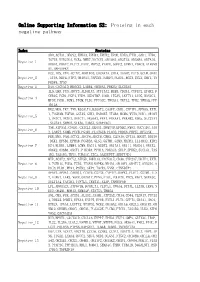
Online Supporting Information S2: Proteins in Each Negative Pathway
Online Supporting Information S2: Proteins in each negative pathway Index Proteins ADO,ACTA1,DEGS2,EPHA3,EPHB4,EPHX2,EPOR,EREG,FTH1,GAD1,HTR6, IGF1R,KIR2DL4,NCR3,NME7,NOTCH1,OR10S1,OR2T33,OR56B4,OR7A10, Negative_1 OR8G1,PDGFC,PLCZ1,PROC,PRPS2,PTAFR,SGPP2,STMN1,VDAC3,ATP6V0 A1,MAPKAPK2 DCC,IDS,VTN,ACTN2,AKR1B10,CACNA1A,CHIA,DAAM2,FUT5,GCLM,GNAZ Negative_2 ,ITPA,NEU4,NTF3,OR10A3,PAPSS1,PARD3,PLOD1,RGS3,SCLY,SHC1,TN FRSF4,TP53 Negative_3 DAO,CACNA1D,HMGCS2,LAMB4,OR56A3,PRKCQ,SLC25A5 IL5,LHB,PGD,ADCY3,ALDH1A3,ATP13A2,BUB3,CD244,CYFIP2,EPHX2,F CER1G,FGD1,FGF4,FZD9,HSD17B7,IL6R,ITGAV,LEFTY1,LIPG,MAN1C1, Negative_4 MPDZ,PGM1,PGM3,PIGM,PLD1,PPP3CC,TBXAS1,TKTL2,TPH2,YWHAQ,PPP 1R12A HK2,MOS,TKT,TNN,B3GALT4,B3GAT3,CASP7,CDH1,CYFIP1,EFNA5,EXTL 1,FCGR3B,FGF20,GSTA5,GUK1,HSD3B7,ITGB4,MCM6,MYH3,NOD1,OR10H Negative_5 1,OR1C1,OR1E1,OR4C11,OR56A3,PPA1,PRKAA1,PRKAB2,RDH5,SLC27A1 ,SLC2A4,SMPD2,STK36,THBS1,SERPINC1 TNR,ATP5A1,CNGB1,CX3CL1,DEGS1,DNMT3B,EFNB2,FMO2,GUCY1B3,JAG Negative_6 2,LARS2,NUMB,PCCB,PGAM1,PLA2G1B,PLOD2,PRDX6,PRPS1,RFXANK FER,MVD,PAH,ACTC1,ADCY4,ADCY8,CBR3,CLDN16,CPT1A,DDOST,DDX56 ,DKK1,EFNB1,EPHA8,FCGR3A,GLS2,GSTM1,GZMB,HADHA,IL13RA2,KIR2 Negative_7 DS4,KLRK1,LAMB4,LGMN,MAGI1,NUDT2,OR13A1,OR1I1,OR4D11,OR4X2, OR6K2,OR8B4,OXCT1,PIK3R4,PPM1A,PRKAG3,SELP,SPHK2,SUCLG1,TAS 1R2,TAS1R3,THY1,TUBA1C,ZIC2,AASDHPPT,SERPIND1 MTR,ACAT2,ADCY2,ATP5D,BMPR1A,CACNA1E,CD38,CYP2A7,DDIT4,EXTL Negative_8 1,FCER1G,FGD3,FZD5,ITGAM,MAPK8,NR4A1,OR10V1,OR4F17,OR52D1,O R8J3,PLD1,PPA1,PSEN2,SKP1,TACR3,VNN1,CTNNBIP1 APAF1,APOA1,CARD11,CCDC6,CSF3R,CYP4F2,DAPK1,FLOT1,GSTM1,IL2 -

Investigating Olfactory Gene Variation and Odour Identification in Older
G C A T T A C G G C A T genes Article Investigating Olfactory Gene Variation and Odour Identification in Older Adults Siddharth Raj 1 , Anbupalam Thalamuthu 1 , Nicola J Armstrong 2 , Margaret J Wright 3,4, John B Kwok 5, Julian N Trollor 1,6 , David Ames 7,8, Peter R Schofield 9,10 , Henry Brodaty 1,11 , Perminder S Sachdev 1,12 and Karen A Mather 1,9,* 1 Centre for Healthy Brain Ageing, Faculty of Medicine, School of Psychiatry, University of New South Wales (UNSW), Sydney, NSW 2031, Australia; [email protected] (S.R.); [email protected] (A.T.); [email protected] (J.N.T.); [email protected] (H.B.); [email protected] (P.S.S.) 2 Department of Mathematics and Statistics, Curtin University, Perth, WA 6102, Australia; [email protected] 3 Queensland Brain Institute, University of Queensland, St. Lucia, QLD 4072, Australia; [email protected] 4 Centre for Advanced Imaging, University of Queensland, St. Lucia, QLD 4072, Australia 5 School of Medical Sciences, University of Sydney, Sydney, NSW 2006, Australia; [email protected] 6 Department of Developmental Disability Neuropsychiatry, UNSW, Sydney, NSW 2031, Australia 7 National Ageing Research Institute, Parkville, VIC 3052, Australia; [email protected] 8 Academic Unit for Psychiatry of Old Age, University of Melbourne, St George’s Hospital, Kew, Melbourne, VIC 3010, Australia 9 Neuroscience Research Australia, Sydney, NSW 2031, Australia; p.schofi[email protected] 10 School of Medical Sciences, UNSW, Sydney, NSW 2031, Australia 11 Dementia Collaborative Research Centre Assessment and Better Care, UNSW, Sydney, NSW 2031, Australia 12 Neuropsychiatric Institute, the Prince of Wales Hospital, Sydney, NSW 2031, Australia * Correspondence: [email protected]; Tel.: +61-(2)-9065-1347 Citation: Raj, S.; Thalamuthu, A.; Armstrong, N.J.; Wright, M.J.; Abstract: Ageing is associated with a decrease in odour identification. -
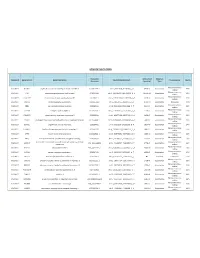
Somatic Mutations
SOMATIC MUTATIONS Transcript Amino Acid Mutation Sample ID Gene Symbol Gene Description Nucleotide (genomic) Consequence Mut % Accession (protein) Type Nonsynonymous PGDX11T ACSBG2 acyl-CoA synthetase bubblegum family member 2 CCDS12159.1 chr19_6141628_6141628_C_A 654A>D Substitution 46% coding Nonsynonymous PGDX11T ATR ataxia telangiectasia and Rad3 related CCDS3124.1 chr3_143721232_143721232_G_A 1451R>W Substitution 32% coding Nonsynonymous PGDX11T C1orf183 chromosome 1 open reading frame 183 CCDS841.1 chr1_112071336_112071336_C_T 224R>Q Substitution 22% coding PGDX11T CMYA5 cardiomyopathy associated 5 NM_153610 chr5_79063455_79063455_C_A 1037C>X Substitution Nonsense 34% Nonsynonymous PGDX11T CNR1 cannabinoid receptor 1 (brain) CCDS5015.1 chr6_88911646_88911646_C_T 23V>M Substitution 35% coding Nonsynonymous PGDX11T COL4A4 collagen; type IV; alpha 4 CCDS42828.1 chr2_227681807_227681807_G_A 227R>C Substitution 20% coding Nonsynonymous PGDX11T CYBASC3 cytochrome b; ascorbate dependent 3 CCDS8004.1 chr11_60877136_60877136_G_A 149R>C Substitution 34% coding Nonsynonymous PGDX11T DYRK3 dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 3 CCDS30999.1 chr1_204888091_204888091_G_A 309V>I Substitution 44% coding Nonsynonymous PGDX11T ELMO1 engulfment and cell motility 1 CCDS5449.1 chr7_37239295_37239295_G_A 160T>M Substitution 24% coding Nonsynonymous PGDX11T FAM83H family with sequence similarity 83; member H CCDS6410.2 chr8_144884473_144884473_C_A 90G>C Substitution 53% coding Nonsynonymous PGDX11T KPTN kaptin (actin binding protein) -

Research/0018.1
http://genomebiology.com/2001/2/6/research/0018.1 Research The human olfactory receptor repertoire comment Sergey Zozulya, ernando Echeverri and Trieu Nguyen Address: Senomyx Inc., 11099 North Torrey Pines Road, La Jolla, CA 92037, USA. Correspondence: Sergey Zozulya. E-mail: [email protected] reviews Published: 1 June 2001 Received: 8 March 2001 Revised: 12 April 2001 Genome Biology 2001, 2(6):research0018.1–0018.12 Accepted: 18 April 2001 The electronic version of this article is the complete one and can be found online at http://genomebiology.com/2001/2/6/research/0018 © 2001 Zozulya et al., licensee BioMed Central Ltd (Print ISSN 1465-6906; Online ISSN 1465-6914) reports Abstract Background: The mammalian olfactory apparatus is able to recognize and distinguish thousands of structurally diverse volatile chemicals. This chemosensory function is mediated by a very large family of seven-transmembrane olfactory (odorant) receptors encoded by approximately 1,000 genes, the majority of which are believed to be pseudogenes in humans. deposited research Results: The strategy of our sequence database mining for full-length, functional candidate odorant receptor genes was based on the high overall sequence similarity and presence of a number of conserved sequence motifs in all known mammalian odorant receptors as well as the absence of introns in their coding sequences. We report here the identification and physical cloning of 347 putative human full-length odorant receptor genes. Comparative sequence analysis of the predicted gene products allowed us to identify and define a number of consensus sequence motifs and structural features of this vast family of receptors.