Cold Exposure Causes Cell Death by Depolarization-Mediated Ca2+
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Glossary - Cellbiology
1 Glossary - Cellbiology Blotting: (Blot Analysis) Widely used biochemical technique for detecting the presence of specific macromolecules (proteins, mRNAs, or DNA sequences) in a mixture. A sample first is separated on an agarose or polyacrylamide gel usually under denaturing conditions; the separated components are transferred (blotting) to a nitrocellulose sheet, which is exposed to a radiolabeled molecule that specifically binds to the macromolecule of interest, and then subjected to autoradiography. Northern B.: mRNAs are detected with a complementary DNA; Southern B.: DNA restriction fragments are detected with complementary nucleotide sequences; Western B.: Proteins are detected by specific antibodies. Cell: The fundamental unit of living organisms. Cells are bounded by a lipid-containing plasma membrane, containing the central nucleus, and the cytoplasm. Cells are generally capable of independent reproduction. More complex cells like Eukaryotes have various compartments (organelles) where special tasks essential for the survival of the cell take place. Cytoplasm: Viscous contents of a cell that are contained within the plasma membrane but, in eukaryotic cells, outside the nucleus. The part of the cytoplasm not contained in any organelle is called the Cytosol. Cytoskeleton: (Gk. ) Three dimensional network of fibrous elements, allowing precisely regulated movements of cell parts, transport organelles, and help to maintain a cell’s shape. • Actin filament: (Microfilaments) Ubiquitous eukaryotic cytoskeletal proteins (one end is attached to the cell-cortex) of two “twisted“ actin monomers; are important in the structural support and movement of cells. Each actin filament (F-actin) consists of two strands of globular subunits (G-Actin) wrapped around each other to form a polarized unit (high ionic cytoplasm lead to the formation of AF, whereas low ion-concentration disassembles AF). -

Interplay Between Gating and Block of Ligand-Gated Ion Channels
brain sciences Review Interplay between Gating and Block of Ligand-Gated Ion Channels Matthew B. Phillips 1,2, Aparna Nigam 1 and Jon W. Johnson 1,2,* 1 Department of Neuroscience, University of Pittsburgh, Pittsburgh, PA 15260, USA; [email protected] (M.B.P.); [email protected] (A.N.) 2 Center for Neuroscience, University of Pittsburgh, Pittsburgh, PA 15260, USA * Correspondence: [email protected]; Tel.: +1-(412)-624-4295 Received: 27 October 2020; Accepted: 26 November 2020; Published: 1 December 2020 Abstract: Drugs that inhibit ion channel function by binding in the channel and preventing current flow, known as channel blockers, can be used as powerful tools for analysis of channel properties. Channel blockers are used to probe both the sophisticated structure and basic biophysical properties of ion channels. Gating, the mechanism that controls the opening and closing of ion channels, can be profoundly influenced by channel blocking drugs. Channel block and gating are reciprocally connected; gating controls access of channel blockers to their binding sites, and channel-blocking drugs can have profound and diverse effects on the rates of gating transitions and on the stability of channel open and closed states. This review synthesizes knowledge of the inherent intertwining of block and gating of excitatory ligand-gated ion channels, with a focus on the utility of channel blockers as analytic probes of ionotropic glutamate receptor channel function. Keywords: ligand-gated ion channel; channel block; channel gating; nicotinic acetylcholine receptor; ionotropic glutamate receptor; AMPA receptor; kainate receptor; NMDA receptor 1. Introduction Neuronal information processing depends on the distribution and properties of the ion channels found in neuronal membranes. -

Effect of Hyperkalemia on Membrane Potential: Depolarization
❖ CASE 3 A 6-year-old boy is brought to the family physician after his parents noticed that he had difficulty moving his arms and legs after a soccer game. About 10 minutes after leaving the field, the boy became so weak that he could not stand for about 30 minutes. Questioning revealed that he had complained of weakness after eating bananas, had frequent muscle spasms, and occasionally had myotonia, which was expressed as difficulty in releasing his grip or diffi- culty opening his eyes after squinting into the sun. After a thorough physical examination, the boy was diagnosed with hyperkalemic periodic paralysis. The family was advised to feed the boy carbohydrate-rich, low-potassium foods, give him glucose-containing drinks during attacks, and have him avoid strenuous exercise and fasting. ◆ What is the effect of hyperkalemia on cell membrane potential? ◆ What is responsible for the repolarizing phase of an action potential? ◆ What is the effect of prolonged depolarization on the skeletal muscle Na+ channel? 32 CASE FILES: PHYSIOLOGY ANSWERS TO CASE 3: ACTION POTENTIAL Summary: A 6-year-old boy who experiences profound weakness after exer- cise is diagnosed with hyperkalemic periodic paralysis. ◆ Effect of hyperkalemia on membrane potential: Depolarization. ◆ Repolarization mechanisms: Activation of voltage-gated K+ conductance and inactivation of Na+ conductance. ◆ Effect of prolonged depolarization: Inactivation of Na+ channels. CLINICAL CORRELATION Hyperkalemic periodic paralysis (HyperPP) is a dominant inherited trait caused by a mutation in the α subunit of the skeletal muscle Na+ channel. It occurs in approximately 1 in 100,000 people and is more common and more severe in males. -

Nicotinic Acetylcholine Receptor Signaling in Neuroprotection
Akinori Akaike · Shun Shimohama Yoshimi Misu Editors Nicotinic Acetylcholine Receptor Signaling in Neuroprotection Nicotinic Acetylcholine Receptor Signaling in Neuroprotection Akinori Akaike • Shun Shimohama Yoshimi Misu Editors Nicotinic Acetylcholine Receptor Signaling in Neuroprotection Editors Akinori Akaike Shun Shimohama Department of Pharmacology, Graduate Department of Neurology, School of School of Pharmaceutical Sciences Medicine Kyoto University Sapporo Medical University Kyoto, Japan Sapporo, Hokkaido, Japan Wakayama Medical University Wakayama, Japan Yoshimi Misu Graduate School of Medicine Yokohama City University Yokohama, Kanagawa, Japan ISBN 978-981-10-8487-4 ISBN 978-981-10-8488-1 (eBook) https://doi.org/10.1007/978-981-10-8488-1 Library of Congress Control Number: 2018936753 © The Editor(s) (if applicable) and The Author(s) 2018. This book is an open access publication. Open Access This book is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made. The images or other third party material in this book are included in the book’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the book’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. The use of general descriptive names, registered names, trademarks, service marks, etc. -

Information Processing in the Axon Dominique Debanne
Information processing in the axon Dominique Debanne To cite this version: Dominique Debanne. Information processing in the axon. Nature Reviews Neuroscience, Nature Publishing Group, 2004, 5 (4), pp.304-316. 10.1038/nrn1397. hal-01766862 HAL Id: hal-01766862 https://hal-amu.archives-ouvertes.fr/hal-01766862 Submitted on 27 Aug 2018 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. INFORMATION PROCESSING IN THE AXON Dominique Debanne Axons link distant brain regions and are generally regarded as reliable transmission cables in which stable propagation occurs once an action potential has been generated. However, recent experimental and theoretical data indicate that the functional capabilities of axons are much more diverse than traditionally thought. Beyond axonal propagation, intrinsic voltage-gated conductances together with the intrinsic geometrical properties of the axon determine complex phenomena such as branch-point failures and reflected propagation. This review considers recent evidence for the role of these forms of axonal computation in the short-term dynamics of neural communication. GAP JUNCTIONS Since the pioneering work of Santiago Ramón y Cajal, of propagation and how intrinsic channels that are Morphological equivalent of the axon has been defined as a long neuronal process present in the axon shape the action potential. -

9.01 Introduction to Neuroscience Fall 2007
MIT OpenCourseWare http://ocw.mit.edu 9.01 Introduction to Neuroscience Fall 2007 For information about citing these materials or our Terms of Use, visit: http://ocw.mit.edu/terms. 9.01 Recitation (R02) RECITATION #2: Tuesday, September 18th Review of Lectures: 3, 4 Reading: Chapters 3, 4 or Neuroscience: Exploring the Brain (3rd edition) Outline of Recitation: I. Previous Recitation: a. Questions on practice exam questions from last recitation? II. Review of Material: a. Exploiting Axoplasmic Transport b. Types of Glia c. THE RESTING MEMBRANE POTENTIAL d. THE ACTION POTENTIAL III. Practice Exam Questions IV. Questions on Pset? Exploiting Axoplasmic Transport: Maps connections of the brain Rates of transport: - slow: - fast: Examples: Uses anterograde transport: - Uses retrograde transport: - - - Types of Glia: - Microglia: - Astrocytes: - Myelinating Glia: 1 THE RESTING MEMBRANE POTENTIAL: The Cast of Chemicals: The Movement of Ions: Influences by two factors: (1) Diffusion: (2) Electricity: Ohm’s Law: I = gV Ionic Equilibrium Potentials (EION): + Example: ENs * diffusional and electrical forces are equal 2 Nernst Equation: EION = 2.303 RT/zF log [ion]0/[ion]i Calculates equilibrium potential for a SINGLE ion. Inside Outside EION (at 37°C) + [K ] + [Na ] 2+ [Ca ] [Cl ] *Pumps maintain concentration gradients (ex. sodiumpotassium pump; calcium pump) Resting Membrane Potential (VM at rest): Measured resting membrane potential: 65 mV Goldman Equation: + + + + VM = 61.54 mV log (PK[K ]o + PNa[Na ]o)/ (PK[K ]I + PNa[Na ]i) + + Calculates membrane potential when permeable to both Na and K . Remember: at REST, gK >>> gNa therefore, VM is closer to EK THE ACTION POTENTIAL (Nerve Impulse): Phases of an Action Potential: Vm (mV) ENa 0 EK Time 3 Conductance of Ion Channels during AP: Remember: Changes in conductance, or permeability of the membrane to a specific ion, changes the membrane potential. -

11 Introduction to the Nervous System and Nervous Tissue
11 Introduction to the Nervous System and Nervous Tissue ou can’t turn on the television or radio, much less go online, without seeing some- 11.1 Overview of the Nervous thing to remind you of the nervous system. From advertisements for medications System 381 Yto treat depression and other psychiatric conditions to stories about celebrities and 11.2 Nervous Tissue 384 their battles with illegal drugs, information about the nervous system is everywhere in 11.3 Electrophysiology our popular culture. And there is good reason for this—the nervous system controls our of Neurons 393 perception and experience of the world. In addition, it directs voluntary movement, and 11.4 Neuronal Synapses 406 is the seat of our consciousness, personality, and learning and memory. Along with the 11.5 Neurotransmitters 413 endocrine system, the nervous system regulates many aspects of homeostasis, including 11.6 Functional Groups respiratory rate, blood pressure, body temperature, the sleep/wake cycle, and blood pH. of Neurons 417 In this chapter we introduce the multitasking nervous system and its basic functions and divisions. We then examine the structure and physiology of the main tissue of the nervous system: nervous tissue. As you read, notice that many of the same principles you discovered in the muscle tissue chapter (see Chapter 10) apply here as well. MODULE 11.1 Overview of the Nervous System Learning Outcomes 1. Describe the major functions of the nervous system. 2. Describe the structures and basic functions of each organ of the central and peripheral nervous systems. 3. Explain the major differences between the two functional divisions of the peripheral nervous system. -

The Nervous System Action Potentials
The Nervous System Action potentials Jennifer Carbrey Ph.D. Department of Cell Biology Nervous System Cell types neurons glial cells Methods of communication in nervous system – action potentials How the nervous system is organized central vs. peripheral Voltage Gated Na+ & K+ Channels image by Rick Melges, Duke University Depolarization of the membrane is the stimulus which leads to both channels opening. To reset the Na+ channel from inactive to closed need to repolarize the membrane. Refractory period is when Na+ channels are inactivated. Action Potentials stage are rapid, “all or none” and do not decay over 1 2 3 4 5 distances top image by image by Chris73 (modified), http://commons.wikimedia.org/wiki/File:Action_potential_%28no_labels%29.svg, Creative Commons Attribution-Share Alike 3.0 Unported license bottom image by Rick Melges, Duke University Unidirectional Propagation of AP image by Rick Melges, Duke University Action potentials move one-way along the axon because of the absolute refractory period of the voltage gated Na+ channel. Integration of Signals Input: Dendrites: ligand gated ion channels, some voltage gated channels; graded potentials Cell body (Soma): ligand gated ion channels; graded potentials Output: Axon: voltage gated ion channels, action potentials Axon initial segment: highest density of voltage gated ion channels & lowest threshold for initiating an action potential, “integrative zone” Axon Initial Segment image by Rick Melges, Duke University Integration of signals at initial segment Saltatory Conduction Node of Ranvier Na+ action potential Large diameter, myelinated axons transmit action potentials very rapidly. Voltage gated channels are concentrated at the nodes. Inactivation of voltage gated Na+ channels insures uni-directional propagation along the axon. -

Responses of Bipolar Cells Each Bipolar Cell Receives Its Direct Input Either from Rods Or Cones
Transduction and Transmission in the Retina 423 Free vesicle FIGURE 20.18 Molecular Components of Tethered vesicle Ribbon Synapses. The electron-dense body of the ribbon is made up largely of the Ribeye (CtBP2) protein, with contributions from CtBP1/ BARS and KIF3A. Piccolo and Rab3-interact- ing molecule 1 (RIM1) are ribbon-associated Ribeye Piccolo proteins. L-type calcium channels cluster in RIM1 the plasma membrane beneath the ribbon and associate with Bassoon, RIM2, Munc13-1, and Presynaptic RIM2 membrane ERC2/CAST1 proteins. Metabotropic (mGluR) Bassoon and ionotropic (iGluR) glutamate receptors Munc13-1 are located in the postsynaptic membranes of bipolar and horizontal cell. (After Dieck and ERC2/CAST1 Brandstatter, 2006.) Ca2+ channel α mGluR 1 subunit iGluR Postsynaptic membranes long before intracellular electrodes were inserted into individual retinal neurons. Although a product of necessity, beginning with the output signal was nonetheless an excellent start- ing point for analyzing retinal processing. Two important features were established by such recordings. The first was that the receptive field of a retinal ganglion cell is organized in a circular, center-surround manner. That is, light in the receptive field surround area produces an opposite effect to that in the center (see Figure 2.5). The second essential feature obtained from ganglion cell recordings was that some retinal ganglion cells were excited by light in their receptive field center, the on retinal ganglion cells; in contrast, other ganglion cells were inhibited by light in their receptive field center, the off retinal ganglion cells. This dichotomy of on and off pathways is now known to be a fundamental property of all vertebrate retinas and establishes an organizing principle for higher visual centers.56 From these recordings of retinal ganglion cells, it is clear that the eye tells the brain about patterns of light and dark. -

Membrane Potentials As Signals
Membrane Potentials as Signals The membrane potential allows a cell to function as a battery, providing electrical power to activities within the cell and between cells. KEY POINTS A membrane potential is the difference in electrical potential between the interior and the exterior of a biological cell. In electrically excitable cells, changes in membrane potential are used for transmitting signals within the cell. The opening and closing of ion channels can induce changes from the resting potential. Depolarization is when the interior voltage becomes more positive and hyperpolarization when it becomes more negative. TERMS Graded potentials Graded potentials vary in size and arise from the summation of the individual actions of ligand- gated ion channel proteins, and decrease over time and space. Depolarization Depolarization is a change in a cell's membrane potential, making it more positive, or less negative, and may result in generation of an action potential. Action potential A short term change in the electrical potential that travels along a cell such as a nerve or muscle fiber. EXAMPLE In neurons, a sufficiently large depolarization can evoke an action potential in which the membrane potential changes rapidly. Give us feedback on this content: Membrane potential (also transmembrane potential or membrane voltage) is the difference in electrical potential between the interior and the exterior of a biological cell. All animal cells are surrounded by a plasma membrane composed of a lipid bilayer with a variety of types of proteins embedded in it. The membrane serves as both an insulator and a diffusion barrier to the movement of ions. -
![Arxiv:1708.00534V1 [Q-Bio.NC] 1 Aug 2017 Myelin and Saltatory Conduction](https://docslib.b-cdn.net/cover/0079/arxiv-1708-00534v1-q-bio-nc-1-aug-2017-myelin-and-saltatory-conduction-2030079.webp)
Arxiv:1708.00534V1 [Q-Bio.NC] 1 Aug 2017 Myelin and Saltatory Conduction
Myelin and saltatory conduction Maurizio De Pitt`a The University of Chicago, USA EPI BEAGLE, INRIA Rhˆone-Alpes, France August 3, 2017 (Submitted as contributed section to the chapter on “Neurophysiology” of the book “Da˜no cerebral” (Brain damage), JC Arango-Asprilla & L Olabarrieta-Landa eds., Manual Moderno Editions.) arXiv:1708.00534v1 [q-bio.NC] 1 Aug 2017 1 Myelin allows fast and reliable conduction of nerve pulses Myelin is a fatty substance that ensheathes the axon of some nerve cells, forming an electrically insulating layer. It is considered a defining characteristic of jawed vertebrates and is essential for the proper functioning of the nervous system of these latter. Myelin is made by different cell types, and varies in chemical composition and configuration, but performs the same insulating function. Myelinated axons look like strings of sausages under a microscope, and because of their white appearance they are integral components of the “white matter” of the brain. The myelin sausages ensue from wrapping of axons by myelin in multiple, concentric layers and are separated from each others by small unmyelinated axonal segments known as nodes of Ranvier. Typically the length of a node is very small (0.1%) compared to the length of the myelinated segment. Single myelinated fibers range in diameter from 0.2 to 20 µm on average, while unmyelinated fibers range between 0.1 and 1 µm [1]. Peripheral axons are myelinated only if their diameter is larger than about 1 µm, and the axonal caliber maintains a rather constant ratio to the myelin sheath thickness [2]. A normal myelin ensheathing of a mature peripheral nerve is usually 100 times as long as the diameter of the corresponding axon [3]. -
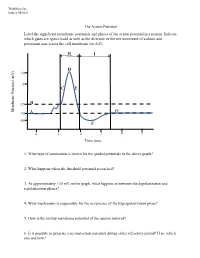
The Action Potential Label the Significant Membrane Potentials and Phases of the Action Potential in a Neuron
Worksheet by Andrey Michel The Action Potential Label the significant membrane potentials and phases of the action potential in a neuron. Indicate which gates are open/closed as well as the direction of the net movement of sodium and potassium ions across the cell membrane for A-G. H I D +30 0 C E -55 B Membrane Potential Membrane (mV) G -70 A -80 F 0 1 2 3 4 5 Time (ms) 1. What type of summation is shown for the graded potentials in the above graph? 2. What happens when the threshold potential is reached? 3. At approximately +30 mV on the graph, what happens in between the depolarization and repolarization phases? 4. What mechanism is responsible for the occurrence of the hyperpolarization phase? 5. How is the resting membrane potential of the neuron restored? 6. Is it possible to generate a second action potential during either refractory period? If so, which one and how? Place the following events in chronological order from 1-8: Na+ enters the cell, and depolarization occurs to approximately +30 mV. The voltage across the cell membrane is -70 mV, the resting membrane potential. Upon reaching the peak of the action potential, the VG Na+ channels are inactivated by the closing of their inactivation gate and the activation gate of each VG K+ channel opens. VG K+ channels close by the closing of their activation gate, and the resting membrane potential is gradually restored. An excitatory post-synaptic potential depolarizes the membrane to threshold and the activation gate of VG Na+ channels open.