4059.Full.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

YES1 Amplification Is a Mechanism of Acquired Resistance to EGFR Inhibitors Identified by Transposon Mutagenesis and Clinical Genomics
YES1 amplification is a mechanism of acquired resistance to EGFR inhibitors identified by transposon mutagenesis and clinical genomics Pang-Dian Fana,b,1, Giuseppe Narzisic, Anitha D. Jayaprakashd,2, Elisa Venturinie,3, Nicolas Robinec, Peter Smibertd, Soren Germerf, Helena A. Yug, Emmet J. Jordang,4, Paul K. Paikg, Yelena Y. Janjigiang, Jamie E. Chaftg, Lu Wanga,5, Achim A. Jungblutha, Sumit Middhaa, Lee Spraggona,b,6, Huan Qiaoh, Christine M. Lovlyh, Mark G. Krisg, Gregory J. Rielyg, Katerina Politii, Harold Varmusj,1,7, and Marc Ladanyia,b,1 aDepartment of Pathology, Memorial Sloan Kettering Cancer Center, New York, NY 10065; bHuman Oncology and Pathogenesis Program, Memorial Sloan Kettering Cancer Center, New York, NY 10065; cComputational Biology, New York Genome Center, New York, NY 10013; dTechnology Innovation Lab, New York Genome Center, New York, NY 10013; eProject Management, New York Genome Center, New York, NY 10013; fSequencing Operations, New York Genome Center, New York, NY 10013; gDivision of Solid Tumor Oncology, Department of Medicine, Memorial Sloan Kettering Cancer Center, New York, NY 10065; hVanderbilt–Ingram Cancer Center, Vanderbilt University School of Medicine, Nashville, TN 37232; iDepartment of Pathology and the Yale Cancer Center, Yale University School of Medicine, New Haven, CT 06520; and jCancer Biology and Genetics Program, Sloan Kettering Institute, Memorial Sloan Kettering Cancer Center, New York, NY 10065 Contributed by Harold Varmus, May 8, 2018 (sent for review October 12, 2017; reviewed by Levi Garraway and Alice T. Shaw) In ∼30% of patients with EGFR-mutant lung adenocarcinomas quired resistance to first-generation EGFR TKIs, the underlying whose disease progresses on EGFR inhibitors, the basis for ac- mechanisms still remain to be identified. -

Oncogenic Inhibition by a Deleted in Liver Cancer Gene Requires Cooperation Between Tensin Binding and Rho-Specific Gtpase-Activating Protein Activities
Oncogenic inhibition by a deleted in liver cancer gene requires cooperation between tensin binding and Rho-specific GTPase-activating protein activities Xiaolan Qian*, Guorong Li*, Holly K. Asmussen*, Laura Asnaghi*, William C. Vass*, Richard Braverman*, Kenneth M. Yamada†, Nicholas C. Popescu‡, Alex G. Papageorge*, and Douglas R. Lowy*§ *Laboratory of Cellular Oncology and ‡Laboratory of Experimental Carcinogenesis, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892; and †Laboratory of Cell and Developmental Biology, National Institute of Dental and Craniofacial Research, National Institutes of Health, Bethesda, MD 20892 Communicated by Ira Pastan, National Institutes of Health, Bethesda, MD, April 2, 2007 (received for review March 1, 2007) The three deleted in liver cancer genes (DLC1–3) encode Rho- The prototypic member, designated DLC1, is localized to GTPase-activating proteins (RhoGAPs) whose expression is fre- chromosome 8p21–22 in a region that is commonly deleted in quently down-regulated or silenced in a variety of human malig- hepatocellular carcinoma (5). Its expression is frequently down- nancies. The RhoGAP activity is required for full DLC-dependent regulated or silenced in various solid tumors and hematologic tumor suppressor activity. Here we report that DLC1 and DLC3 bind malignancies, predominantly by promoter methylation (6–13). to human tensin1 and its chicken homolog. The binding has been Ectopic reexpression in DLC1-deficient cancer cell lines can mapped to the tensin Src homology 2 (SH2) and phosphotyrosine suppress cell proliferation, induce apoptosis, and reduce tumor- binding (PTB) domains at the C terminus of tensin proteins. Distinct igenicity. The RhoGAP activity appears to be required for these DLC1 sequences are required for SH2 and PTB binding. -

Src-Family Kinases Impact Prognosis and Targeted Therapy in Flt3-ITD+ Acute Myeloid Leukemia
Src-Family Kinases Impact Prognosis and Targeted Therapy in Flt3-ITD+ Acute Myeloid Leukemia Title Page by Ravi K. Patel Bachelor of Science, University of Minnesota, 2013 Submitted to the Graduate Faculty of School of Medicine in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Pittsburgh 2019 Commi ttee Membership Pa UNIVERSITY OF PITTSBURGH SCHOOL OF MEDICINE Commi ttee Membership Page This dissertation was presented by Ravi K. Patel It was defended on May 31, 2019 and approved by Qiming (Jane) Wang, Associate Professor Pharmacology and Chemical Biology Vaughn S. Cooper, Professor of Microbiology and Molecular Genetics Adrian Lee, Professor of Pharmacology and Chemical Biology Laura Stabile, Research Associate Professor of Pharmacology and Chemical Biology Thomas E. Smithgall, Dissertation Director, Professor and Chair of Microbiology and Molecular Genetics ii Copyright © by Ravi K. Patel 2019 iii Abstract Src-Family Kinases Play an Important Role in Flt3-ITD Acute Myeloid Leukemia Prognosis and Drug Efficacy Ravi K. Patel, PhD University of Pittsburgh, 2019 Abstract Acute myelogenous leukemia (AML) is a disease characterized by undifferentiated bone-marrow progenitor cells dominating the bone marrow. Currently the five-year survival rate for AML patients is 27.4 percent. Meanwhile the standard of care for most AML patients has not changed for nearly 50 years. We now know that AML is a genetically heterogeneous disease and therefore it is unlikely that all AML patients will respond to therapy the same way. Upregulation of protein-tyrosine kinase signaling pathways is one common feature of some AML tumors, offering opportunities for targeted therapy. -

Anti-Yes1 Picoband Antibody Catalog # ABO12155
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 Anti-Yes1 Picoband Antibody Catalog # ABO12155 Specification Anti-Yes1 Picoband Antibody - Product Information Application WB, IHC Primary Accession P07947 Host Rabbit Reactivity Human Clonality Polyclonal Format Lyophilized Description Rabbit IgG polyclonal antibody for Tyrosine-protein kinase Yes(YES1) detection. Tested with WB, IHC-P in Human. Reconstitution Add 0.2ml of distilled water will yield a concentration of 500ug/ml. Anti- YES1 Picoband antibody, ABO12155, Anti-Yes1 Picoband Antibody - Additional Western blottingAll lanes: Anti YES1 Information (ABO12155) at 0.5ug/mlLane 1: SGC Whole Cell Lysate at 40ugLane 2: A549 Whole Cell Lysate at 40ugLane 3: HEPG2 Whole Cell Gene ID 7525 Lysate at 40ugPredicted bind size: Other Names 61KDObserved bind size: 61KD Tyrosine-protein kinase Yes, 2.7.10.2, Proto-oncogene c-Yes, p61-Yes, YES1, YES Calculated MW 60801 MW KDa Application Details Immunohistochemistry(Paraffin-embedded Section), 0.5-1 µg/ml, Human, By Heat<br>Western blot, 0.1-0.5 µg/ml, Human<br> Subcellular Localization Cell membrane. Cytoplasm, cytoskeleton, microtubule organizing center, centrosome. Anti- YES1 Picoband antibody, Cytoplasm, cytosol. Newly synthesized ABO12155,IHC(P)IHC(P): Human Mammary protein initially accumulates in the Golgi Cancer Tissue region and traffics to the plasma membrane through the exocytic pathway. Anti-Yes1 Picoband Antibody - Background Tissue Specificity Expressed in the epithelial cells of renal Proto-oncogene tyrosine-protein kinase Yes is proximal tubules and stomach as well as an enzyme that in humans is encoded by the Page 1/3 10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 hematopoietic cells in the bone marrow and YES1 gene. -

Transcriptomic and Proteomic Profiling Provides Insight Into
BASIC RESEARCH www.jasn.org Transcriptomic and Proteomic Profiling Provides Insight into Mesangial Cell Function in IgA Nephropathy † † ‡ Peidi Liu,* Emelie Lassén,* Viji Nair, Celine C. Berthier, Miyuki Suguro, Carina Sihlbom,§ † | † Matthias Kretzler, Christer Betsholtz, ¶ Börje Haraldsson,* Wenjun Ju, Kerstin Ebefors,* and Jenny Nyström* *Department of Physiology, Institute of Neuroscience and Physiology, §Proteomics Core Facility at University of Gothenburg, University of Gothenburg, Gothenburg, Sweden; †Division of Nephrology, Department of Internal Medicine and Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, Michigan; ‡Division of Molecular Medicine, Aichi Cancer Center Research Institute, Nagoya, Japan; |Department of Immunology, Genetics and Pathology, Uppsala University, Uppsala, Sweden; and ¶Integrated Cardio Metabolic Centre, Karolinska Institutet Novum, Huddinge, Sweden ABSTRACT IgA nephropathy (IgAN), the most common GN worldwide, is characterized by circulating galactose-deficient IgA (gd-IgA) that forms immune complexes. The immune complexes are deposited in the glomerular mesangium, leading to inflammation and loss of renal function, but the complete pathophysiology of the disease is not understood. Using an integrated global transcriptomic and proteomic profiling approach, we investigated the role of the mesangium in the onset and progression of IgAN. Global gene expression was investigated by microarray analysis of the glomerular compartment of renal biopsy specimens from patients with IgAN (n=19) and controls (n=22). Using curated glomerular cell type–specific genes from the published literature, we found differential expression of a much higher percentage of mesangial cell–positive standard genes than podocyte-positive standard genes in IgAN. Principal coordinate analysis of expression data revealed clear separation of patient and control samples on the basis of mesangial but not podocyte cell–positive standard genes. -

Personalized Prediction of Acquired Resistance to EGFR-Targeted Inhibitors Using a Pathway-Based Machine Learning Approach
Cancers 2019, 11, x S1 of S9 Supplementary Materials: Personalized Prediction of Acquired Resistance to EGFR-Targeted Inhibitors Using a Pathway-Based Machine Learning Approach Young Rae Kim, Yong Wan Kim, Suh Eun Lee, Hye Won Yang and Sung Young Kim Table S1. Characteristics of individual studies. Sample Size Origin of Cancer Drug Dataset Platform S AR (Cell Lines) Lung cancer Agilent-014850 Whole Human GSE34228 26 26 (PC9) Genome Microarray 4x44K Gefitinib Epidermoid carcinoma Affymetrix Human Genome U133 GSE10696 3 3 (A431) Plus 2.0 Head and neck cancer Illumina HumanHT-12 V4.0 GSE62061 12 12 (Cal-27, SSC-25, FaDu, expression beadchip SQ20B) Erlotinib Head and neck cancer Illumina HumanHT-12 V4.0 GSE49135 3 3 (HN5) expression beadchip Lung cancer (HCC827, Illumina HumanHT-12 V3.0 GSE38310 3 6 ER3, T15-2) expression beadchip Lung cancer Illumina HumanHT-12 V3.0 GSE62504 1 2 (HCC827) expression beadchip Afatinib Lung cancer * Illumina HumanHT-12 V4.0 GSE75468 1 3 (HCC827) expression beadchip Head and neck cancer Affymetrix Human Genome U133 Cetuximab GSE21483 3 3 (SCC1) Plus 2.0 Array GEO, gene expression omnibus; GSE, gene expression series; S, sensitive; AR, acquired EGFR-TKI resistant; * Lung Cancer Cells Derived from Tumor Xenograft Model. Table S2. The performances of four penalized regression models. Model ACC precision recall F1 MCC AUROC BRIER Ridge 0.889 0.852 0.958 0.902 0.782 0.964 0.129 Lasso 0.944 0.957 0.938 0.947 0.889 0.991 0.042 Elastic Net 0.978 0.979 0.979 0.979 0.955 0.999 0.023 EPSGO Elastic Net 0.989 1.000 0.979 0.989 0.978 1.000 0.018 AUROC, area under curve of receiver operating characteristic; ACC, accuracy; MCC, Matthews correlation coefficient; EPSGO, Efficient Parameter Selection via Global Optimization algorithm. -

Genetic Alterations of Protein Tyrosine Phosphatases in Human Cancers
Oncogene (2015) 34, 3885–3894 © 2015 Macmillan Publishers Limited All rights reserved 0950-9232/15 www.nature.com/onc REVIEW Genetic alterations of protein tyrosine phosphatases in human cancers S Zhao1,2,3, D Sedwick3,4 and Z Wang2,3 Protein tyrosine phosphatases (PTPs) are enzymes that remove phosphate from tyrosine residues in proteins. Recent whole-exome sequencing of human cancer genomes reveals that many PTPs are frequently mutated in a variety of cancers. Among these mutated PTPs, PTP receptor T (PTPRT) appears to be the most frequently mutated PTP in human cancers. Beside PTPN11, which functions as an oncogene in leukemia, genetic and functional studies indicate that most of mutant PTPs are tumor suppressor genes. Identification of the substrates and corresponding kinases of the mutant PTPs may provide novel therapeutic targets for cancers harboring these mutant PTPs. Oncogene (2015) 34, 3885–3894; doi:10.1038/onc.2014.326; published online 29 September 2014 INTRODUCTION tyrosine/threonine-specific phosphatases. (4) Class IV PTPs include Protein tyrosine phosphorylation has a critical role in virtually all four Drosophila Eya homologs (Eya1, Eya2, Eya3 and Eya4), which human cellular processes that are involved in oncogenesis.1 can dephosphorylate both tyrosine and serine residues. Protein tyrosine phosphorylation is coordinately regulated by protein tyrosine kinases (PTKs) and protein tyrosine phosphatases 1 THE THREE-DIMENSIONAL STRUCTURE AND CATALYTIC (PTPs). Although PTKs add phosphate to tyrosine residues in MECHANISM OF PTPS proteins, PTPs remove it. Many PTKs are well-documented oncogenes.1 Recent cancer genomic studies provided compelling The three-dimensional structures of the catalytic domains of evidence that many PTPs function as tumor suppressor genes, classical PTPs (RPTPs and non-RPTPs) are extremely well because a majority of PTP mutations that have been identified in conserved.5 Even the catalytic domain structures of the dual- human cancers are loss-of-function mutations. -

In This Table Protein Name, Uniprot Code, Gene Name P-Value
Supplementary Table S1: In this table protein name, uniprot code, gene name p-value and Fold change (FC) for each comparison are shown, for 299 of the 301 significantly regulated proteins found in both comparisons (p-value<0.01, fold change (FC) >+/-0.37) ALS versus control and FTLD-U versus control. Two uncharacterized proteins have been excluded from this list Protein name Uniprot Gene name p value FC FTLD-U p value FC ALS FTLD-U ALS Cytochrome b-c1 complex P14927 UQCRB 1.534E-03 -1.591E+00 6.005E-04 -1.639E+00 subunit 7 NADH dehydrogenase O95182 NDUFA7 4.127E-04 -9.471E-01 3.467E-05 -1.643E+00 [ubiquinone] 1 alpha subcomplex subunit 7 NADH dehydrogenase O43678 NDUFA2 3.230E-04 -9.145E-01 2.113E-04 -1.450E+00 [ubiquinone] 1 alpha subcomplex subunit 2 NADH dehydrogenase O43920 NDUFS5 1.769E-04 -8.829E-01 3.235E-05 -1.007E+00 [ubiquinone] iron-sulfur protein 5 ARF GTPase-activating A0A0C4DGN6 GIT1 1.306E-03 -8.810E-01 1.115E-03 -7.228E-01 protein GIT1 Methylglutaconyl-CoA Q13825 AUH 6.097E-04 -7.666E-01 5.619E-06 -1.178E+00 hydratase, mitochondrial ADP/ATP translocase 1 P12235 SLC25A4 6.068E-03 -6.095E-01 3.595E-04 -1.011E+00 MIC J3QTA6 CHCHD6 1.090E-04 -5.913E-01 2.124E-03 -5.948E-01 MIC J3QTA6 CHCHD6 1.090E-04 -5.913E-01 2.124E-03 -5.948E-01 Protein kinase C and casein Q9BY11 PACSIN1 3.837E-03 -5.863E-01 3.680E-06 -1.824E+00 kinase substrate in neurons protein 1 Tubulin polymerization- O94811 TPPP 6.466E-03 -5.755E-01 6.943E-06 -1.169E+00 promoting protein MIC C9JRZ6 CHCHD3 2.912E-02 -6.187E-01 2.195E-03 -9.781E-01 Mitochondrial 2- -

Protein Tyrosine Kinases: Their Roles and Their Targeting in Leukemia
cancers Review Protein Tyrosine Kinases: Their Roles and Their Targeting in Leukemia Kalpana K. Bhanumathy 1,*, Amrutha Balagopal 1, Frederick S. Vizeacoumar 2 , Franco J. Vizeacoumar 1,3, Andrew Freywald 2 and Vincenzo Giambra 4,* 1 Division of Oncology, College of Medicine, University of Saskatchewan, Saskatoon, SK S7N 5E5, Canada; [email protected] (A.B.); [email protected] (F.J.V.) 2 Department of Pathology and Laboratory Medicine, College of Medicine, University of Saskatchewan, Saskatoon, SK S7N 5E5, Canada; [email protected] (F.S.V.); [email protected] (A.F.) 3 Cancer Research Department, Saskatchewan Cancer Agency, 107 Wiggins Road, Saskatoon, SK S7N 5E5, Canada 4 Institute for Stem Cell Biology, Regenerative Medicine and Innovative Therapies (ISBReMIT), Fondazione IRCCS Casa Sollievo della Sofferenza, 71013 San Giovanni Rotondo, FG, Italy * Correspondence: [email protected] (K.K.B.); [email protected] (V.G.); Tel.: +1-(306)-716-7456 (K.K.B.); +39-0882-416574 (V.G.) Simple Summary: Protein phosphorylation is a key regulatory mechanism that controls a wide variety of cellular responses. This process is catalysed by the members of the protein kinase su- perfamily that are classified into two main families based on their ability to phosphorylate either tyrosine or serine and threonine residues in their substrates. Massive research efforts have been invested in dissecting the functions of tyrosine kinases, revealing their importance in the initiation and progression of human malignancies. Based on these investigations, numerous tyrosine kinase inhibitors have been included in clinical protocols and proved to be effective in targeted therapies for various haematological malignancies. -
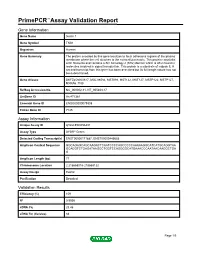
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name tensin 1 Gene Symbol TNS1 Organism Human Gene Summary The protein encoded by this gene localizes to focal adhesions regions of the plasma membrane where the cell attaches to the extracellular matrix. This protein crosslinks actin filaments and contains a Src homology 2 (SH2) domain which is often found in molecules involved in signal transduction. This protein is a substrate of calpain II. A second transcript from this gene has been described but its full length nature has not been determined. Gene Aliases DKFZp586K0617, MGC88584, MST091, MST122, MST127, MSTP122, MSTP127, MXRA6, TNS RefSeq Accession No. NC_000002.11, NT_005403.17 UniGene ID Hs.471381 Ensembl Gene ID ENSG00000079308 Entrez Gene ID 7145 Assay Information Unique Assay ID qHsaCED0056481 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000171887, ENST00000446688 Amplicon Context Sequence GGCAGAGCAGCAAGACTCAATCCCCAGCCCCCAAAAAGGCATCATGCAGGTAA GCAGGTCTGAGATAAGGCTGGTCCAGGGGCATGAAACCCAATAACAAGGCTGA G Amplicon Length (bp) 77 Chromosome Location 2:218668016-218668122 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 109 R2 0.9936 cDNA Cq 23.48 cDNA Tm (Celsius) 83 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq 25.01 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report TNS1, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis -

Nº Ref Uniprot Proteína Péptidos Identificados Por MS/MS 1 P01024
Document downloaded from http://www.elsevier.es, day 26/09/2021. This copy is for personal use. Any transmission of this document by any media or format is strictly prohibited. Nº Ref Uniprot Proteína Péptidos identificados 1 P01024 CO3_HUMAN Complement C3 OS=Homo sapiens GN=C3 PE=1 SV=2 por 162MS/MS 2 P02751 FINC_HUMAN Fibronectin OS=Homo sapiens GN=FN1 PE=1 SV=4 131 3 P01023 A2MG_HUMAN Alpha-2-macroglobulin OS=Homo sapiens GN=A2M PE=1 SV=3 128 4 P0C0L4 CO4A_HUMAN Complement C4-A OS=Homo sapiens GN=C4A PE=1 SV=1 95 5 P04275 VWF_HUMAN von Willebrand factor OS=Homo sapiens GN=VWF PE=1 SV=4 81 6 P02675 FIBB_HUMAN Fibrinogen beta chain OS=Homo sapiens GN=FGB PE=1 SV=2 78 7 P01031 CO5_HUMAN Complement C5 OS=Homo sapiens GN=C5 PE=1 SV=4 66 8 P02768 ALBU_HUMAN Serum albumin OS=Homo sapiens GN=ALB PE=1 SV=2 66 9 P00450 CERU_HUMAN Ceruloplasmin OS=Homo sapiens GN=CP PE=1 SV=1 64 10 P02671 FIBA_HUMAN Fibrinogen alpha chain OS=Homo sapiens GN=FGA PE=1 SV=2 58 11 P08603 CFAH_HUMAN Complement factor H OS=Homo sapiens GN=CFH PE=1 SV=4 56 12 P02787 TRFE_HUMAN Serotransferrin OS=Homo sapiens GN=TF PE=1 SV=3 54 13 P00747 PLMN_HUMAN Plasminogen OS=Homo sapiens GN=PLG PE=1 SV=2 48 14 P02679 FIBG_HUMAN Fibrinogen gamma chain OS=Homo sapiens GN=FGG PE=1 SV=3 47 15 P01871 IGHM_HUMAN Ig mu chain C region OS=Homo sapiens GN=IGHM PE=1 SV=3 41 16 P04003 C4BPA_HUMAN C4b-binding protein alpha chain OS=Homo sapiens GN=C4BPA PE=1 SV=2 37 17 Q9Y6R7 FCGBP_HUMAN IgGFc-binding protein OS=Homo sapiens GN=FCGBP PE=1 SV=3 30 18 O43866 CD5L_HUMAN CD5 antigen-like OS=Homo -

Discovery and Systematic Characterization of Risk Variants and Genes For
medRxiv preprint doi: https://doi.org/10.1101/2021.05.24.21257377; this version posted June 2, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY 4.0 International license . 1 Discovery and systematic characterization of risk variants and genes for 2 coronary artery disease in over a million participants 3 4 Krishna G Aragam1,2,3,4*, Tao Jiang5*, Anuj Goel6,7*, Stavroula Kanoni8*, Brooke N Wolford9*, 5 Elle M Weeks4, Minxian Wang3,4, George Hindy10, Wei Zhou4,11,12,9, Christopher Grace6,7, 6 Carolina Roselli3, Nicholas A Marston13, Frederick K Kamanu13, Ida Surakka14, Loreto Muñoz 7 Venegas15,16, Paul Sherliker17, Satoshi Koyama18, Kazuyoshi Ishigaki19, Bjørn O Åsvold20,21,22, 8 Michael R Brown23, Ben Brumpton20,21, Paul S de Vries23, Olga Giannakopoulou8, Panagiota 9 Giardoglou24, Daniel F Gudbjartsson25,26, Ulrich Güldener27, Syed M. Ijlal Haider15, Anna 10 Helgadottir25, Maysson Ibrahim28, Adnan Kastrati27,29, Thorsten Kessler27,29, Ling Li27, Lijiang 11 Ma30,31, Thomas Meitinger32,33,29, Sören Mucha15, Matthias Munz15, Federico Murgia28, Jonas B 12 Nielsen34,20, Markus M Nöthen35, Shichao Pang27, Tobias Reinberger15, Gudmar Thorleifsson25, 13 Moritz von Scheidt27,29, Jacob K Ulirsch4,11,36, EPIC-CVD Consortium, Biobank Japan, David O 14 Arnar25,37,38, Deepak S Atri39,3, Noël P Burtt4, Maria C Costanzo4, Jason Flannick40, Rajat M 15 Gupta39,3,4, Kaoru Ito18, Dong-Keun Jang4,