Hexachlorophene: Its Absorption, Distribution
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

National Center for Toxicological Research
National Center for Toxicological Research Annual Report Research Accomplishments and Plans FY 2015 – FY 2016 Page 0 of 193 Table of Contents Preface – William Slikker, Jr., Ph.D. ................................................................................... 3 NCTR Vision ......................................................................................................................... 7 NCTR Mission ...................................................................................................................... 7 NCTR Strategic Plan ............................................................................................................ 7 NCTR Organizational Structure .......................................................................................... 8 NCTR Location and Facilities .............................................................................................. 9 NCTR Advances Research Through Outreach and Collaboration ................................... 10 NCTR Global Outreach and Training Activities ............................................................... 12 Global Summit on Regulatory Science .................................................................................................12 Training Activities .................................................................................................................................14 NCTR Scientists – Leaders in the Research Community .................................................. 15 Science Advisory Board ................................................................................................... -

A Screening-Based Approach to Circumvent Tumor Microenvironment
JBXXXX10.1177/1087057113501081Journal of Biomolecular ScreeningSingh et al. 501081research-article2013 Original Research Journal of Biomolecular Screening 2014, Vol 19(1) 158 –167 A Screening-Based Approach to © 2013 Society for Laboratory Automation and Screening DOI: 10.1177/1087057113501081 Circumvent Tumor Microenvironment- jbx.sagepub.com Driven Intrinsic Resistance to BCR-ABL+ Inhibitors in Ph+ Acute Lymphoblastic Leukemia Harpreet Singh1,2, Anang A. Shelat3, Amandeep Singh4, Nidal Boulos1, Richard T. Williams1,2*, and R. Kiplin Guy2,3 Abstract Signaling by the BCR-ABL fusion kinase drives Philadelphia chromosome–positive acute lymphoblastic leukemia (Ph+ ALL) and chronic myelogenous leukemia (CML). Despite their clinical activity in many patients with CML, the BCR-ABL kinase inhibitors (BCR-ABL-KIs) imatinib, dasatinib, and nilotinib provide only transient leukemia reduction in patients with Ph+ ALL. While host-derived growth factors in the leukemia microenvironment have been invoked to explain this drug resistance, their relative contribution remains uncertain. Using genetically defined murine Ph+ ALL cells, we identified interleukin 7 (IL-7) as the dominant host factor that attenuates response to BCR-ABL-KIs. To identify potential combination drugs that could overcome this IL-7–dependent BCR-ABL-KI–resistant phenotype, we screened a small-molecule library including Food and Drug Administration–approved drugs. Among the validated hits, the well-tolerated antimalarial drug dihydroartemisinin (DHA) displayed potent activity in vitro and modest in vivo monotherapy activity against engineered murine BCR-ABL-KI–resistant Ph+ ALL. Strikingly, cotreatment with DHA and dasatinib in vivo strongly reduced primary leukemia burden and improved long-term survival in a murine model that faithfully captures the BCR-ABL-KI–resistant phenotype of human Ph+ ALL. -

Avant-Propos Le Format De Présentation De Cette Thèse Correspond À Une Recommandation À La Spécialité
Avant-propos Le format de présentation de cette thèse correspond à une recommandation à la spécialité Maladies infectieuses, à l’intérieur du Master des Sciences de la Vie et de la Santé qui dépend de l’Ecole Doctorale des Sciences de la Vie de Marseille. Le candidat est amené à respecter les règles qui lui sont imposées et qui comportent un format de thèse utilisé dans le Nord de l’Europe et qui permet un meilleur rangement que les thèses traditionnelles. Par ailleurs, la partie introduction et bibliographie est remplacée par une revue envoyée dans un journal afin de permettre une évaluation extérieure de la qualité de la revue et de permettre à l’étudiant de commencer le plus tôt possible une bibliographie sur le domaine de cette thèse. Par ailleurs, la thèse est présentée sur article publié, accepté, ou soumis associé d’un bref commentaire donnant le sens général du travail. Cette forme de présentation a paru plus en adéquation avec les exigences de la compétition internationale et permet de se concentrer sur des travaux qui bénéficieront d’une diffusion internationale. Professeur Didier RAOULT 2 Remerciements J’adresse mes remerciements aux personnes qui ont contribué à la réalisation de ce travail. En premier lieu, au Professeur Didier RALOUT, qui m’a accueillie au sein de l’IHU Méditerranée Infection. Au Professeur Serge MORAND, au Docteur Marie KEMPF et au Professeur Philippe COLSON de m’avoir honorée en acceptant d’être rapporteurs et examinateurs de cette thèse. Je souhaite particulièrement remercier : Le Professeur Jean-Marc ROLAIN, de m’avoir accueillie dans son équipe et m’avoir orientée et soutenue tout au long de ces trois années de thèse. -

Step-By-Step Guide to Better Laboratory Management Practices
Step-by-Step Guide to Better Laboratory Management Practices Prepared by The Washington State Department of Ecology Hazardous Waste and Toxics Reduction Program Publication No. 97- 431 Revised January 2003 Printed on recycled paper For additional copies of this document, contact: Department of Ecology Publications Distribution Center PO Box 47600 Olympia, WA 98504-7600 (360) 407-7472 or 1 (800) 633-7585 or contact your regional office: Department of Ecology’s Regional Offices (425) 649-7000 (509) 575-2490 (509) 329-3400 (360) 407-6300 The Department of Ecology is an equal opportunity agency and does not discriminate on the basis of race, creed, color, disability, age, religion, national origin, sex, marital status, disabled veteran’s status, Vietnam Era veteran’s status or sexual orientation. If you have special accommodation needs, or require this document in an alternate format, contact the Hazardous Waste and Toxics Reduction Program at (360)407-6700 (voice) or 711 or (800) 833-6388 (TTY). Table of Contents Introduction ....................................................................................................................................iii Section 1 Laboratory Hazardous Waste Management ...........................................................1 Designating Dangerous Waste................................................................................................1 Counting Wastes .......................................................................................................................8 Treatment by Generator...........................................................................................................12 -

Chlorhexidine Bathing in a Tertiary Care Neonatal Intensive Care Unit
Chlorhexidine Bathing in a Tertiary Care Neonatal Intensive Care Unit: Impact on Central Line–Associated Bloodstream Infections Author(s): Caroline Quach, MD, MSc; Aaron M. Milstone, MD, MHS; Chantal Perpête, RN, LSH, LSHH; Mario Bonenfant, RN; Dorothy L. Moore, MD, PhD; Therese Perreault, MD Source: Infection Control and Hospital Epidemiology, Vol. 35, No. 2 (February 2014), pp. 158- 163 Published by: The University of Chicago Press on behalf of The Society for Healthcare Epidemiology of America Stable URL: http://www.jstor.org/stable/10.1086/674862 . Accessed: 24/01/2014 14:25 Your use of the JSTOR archive indicates your acceptance of the Terms & Conditions of Use, available at . http://www.jstor.org/page/info/about/policies/terms.jsp . JSTOR is a not-for-profit service that helps scholars, researchers, and students discover, use, and build upon a wide range of content in a trusted digital archive. We use information technology and tools to increase productivity and facilitate new forms of scholarship. For more information about JSTOR, please contact [email protected]. The University of Chicago Press and The Society for Healthcare Epidemiology of America are collaborating with JSTOR to digitize, preserve and extend access to Infection Control and Hospital Epidemiology. http://www.jstor.org This content downloaded from 209.172.182.131 on Fri, 24 Jan 2014 14:25:10 PM All use subject to JSTOR Terms and Conditions infection control and hospital epidemiology february 2014, vol. 35, no. 2 original article Chlorhexidine Bathing in a Tertiary Care Neonatal Intensive Care Unit: Impact on Central Line–Associated Bloodstream Infections Caroline Quach, MD, MSc;1,2,3 Aaron M. -
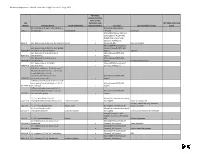
Chemicals of High Concern List (Sorted Alphabetically), July 2010
Minnesota Department of Health, Chemicals of High Concern list, July 1, 2010 Persistent, Bioaccumulative, Toxic or very CAS Persistent, very HPV (2006 and 3 of 4 Number Chemical Name Health endpoint(s) Bioaccumulative Source(s) Use example(s) or class years) (S)-4-hydroxy-3-(3-oxo-1-phenylbutyl)-2- Maine (EU Reproductive 5543-57-7 benzopyrone Reproduction Toxicant) Sunscreen Maine (CA Prop 65; IARC; EU Carcinogen; NTP 11th ROC; OSPAR Chemicals of High Concern); WA Appen1; 91-94-1 [1,1'-biphenyl]-4,4'-diamine, 3,3'-dichloro-Cancer x Minnesota HRL Dye, curing agent Maine (OSPAR Chemicals of [1,1'-biphenyl]-4,4'-diamine, N,N'-bis(2,4- Concern; Canada PBiT); WA 29398-96-7 dinitrophenyl)-3,3'-dimethoxy- x Appen1 Colorant [1,1'-Biphenyl]-4-ol, 3,4',5-tris(1,1- Maine (Canada PBiT); WA 6257-39-2 dimethylethyl)- x Appen1 [1,1'-Biphenyl]-4-ol, 3,4'-bis(1,1- Maine (Canada PBiT); WA 42479-88-9 dimethylethyl)- x Appen1 Chemical intermediate [1,1'-biphenyl]-4-ol, 3,5-bis(1,1- Maine (OSPAR Chemicals of 2668-47-5 dimethylethyl)- x Concern); WA Appen1 [2,6'-Bibenzothiazole]-7-sulfonic acid, 2'- (4-aminophenyl)-6-methyl-, diazotized, coupled with diazotized 4- aminobenzenesulfonic acid and Maine (Canada PBiT); WA 91696-90-1 resorcinol, sodium salts x Appen1 1(2H)-Quinolineethanol, 6-[(2-chloro-4,6- dinitrophenyl) azo]-3,4-dihydro-2,2,4,7- Maine (Canada PBiT); WA 63133-84-6 tetramethyl- x Appen1 1(2H)-Quinolinepropanamide, 6-(2,2- dicyanoethenyl)-3, 4-dihydro-2,2,4,7- Maine (Canada PBiT); WA 63467-15-2 tetramethyl-N-phenyl- x Appen1 1,1,1,2-Tetrachloro-2,2-bis(4- -

Guideline for Disinfection and Sterilization in Healthcare Facilities, 2008
Guideline for Disinfection and Sterilization in Healthcare Facilities, 2008 Guideline for Disinfection and Sterilization in Healthcare Facilities, 2008 William A. Rutala, Ph.D., M.P.H.1,2, David J. Weber, M.D., M.P.H.1,2, and the Healthcare Infection Control Practices Advisory Committee (HICPAC)3 1Hospital Epidemiology University of North Carolina Health Care System Chapel Hill, NC 27514 2Division of Infectious Diseases University of North Carolina School of Medicine Chapel Hill, NC 27599-7030 1 Guideline for Disinfection and Sterilization in Healthcare Facilities, 2008 3HICPAC Members Robert A. Weinstein, MD (Chair) Cook County Hospital Chicago, IL Jane D. Siegel, MD (Co-Chair) University of Texas Southwestern Medical Center Dallas, TX Michele L. Pearson, MD (Executive Secretary) Centers for Disease Control and Prevention Atlanta, GA Raymond Y.W. Chinn, MD Sharp Memorial Hospital San Diego, CA Alfred DeMaria, Jr, MD Massachusetts Department of Public Health Jamaica Plain, MA James T. Lee, MD, PhD University of Minnesota Minneapolis, MN William A. Rutala, PhD, MPH University of North Carolina Health Care System Chapel Hill, NC William E. Scheckler, MD University of Wisconsin Madison, WI Beth H. Stover, RN Kosair Children’s Hospital Louisville, KY Marjorie A. Underwood, RN, BSN CIC Mt. Diablo Medical Center Concord, CA This guideline discusses use of products by healthcare personnel in healthcare settings such as hospitals, ambulatory care and home care; the recommendations are not intended for consumer use of the products discussed. 2 -
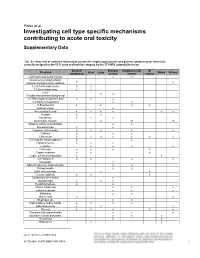
Investigating Cell Type Specific Mechanisms Contributing to Acute Oral Toxicity
Prieto et al.: Investigating cell type specific mechanisms contributing to acute oral toxicity Supplementary Data1 Tab. S1: Overview of collected information on specific target organ/system and general cytotoxicity for chemicals correctly assigned to the CLP acute oral toxicity category by the 3T3 NRU cytotoxicity assay General Nervous Cardiovascular GI Chemical Liver Lung Blood Kidney cytotoxicity system system system (±)-Propranolol hydrochloride x ax (4-ammonio-m-tolyl)ethyl(2- x x hydroxyethyl)ammonium sulphate 1,2,4-Trichlorobenzene x x 1,2-Dichlorobenzene x x 2,4,6- x x Tris(dimethylaminomethyl)phenol 2,4-Dichlorophenoxyacetic acid x x x 5,5-Diphenylhydantoin x x 5-Fluorouracil x x x x Acetophenone x Acetylsalicylic acid x x x x x x Acrolein x x Acrylamide x x x Ammonium chloride x ax ax Atropine sulfate monohydrate x x Benzaldehyde x x Cadmium (III) chloride x x x x x Caffeine x x x Chloroform x x ax x x x x Chloroquine bis(phosphate) x x Chlorpromazine x x x Codeine x x x x Colchicine x x x x Copper sulphate x x x Cupric sulfate pentahydrate x x Cyclosporin A x x x x Diazepam x Diphenhydramine hydrochloride x x Disopyramide x x Ethyl chloroacetate x x Ferrous sulphate x x x Glufosinate-ammonium x Glutethimide x ax x Hexachlorophene x x Lithium Carbonate x x x Lithium sulphate x x x Malathion x x Maleic acid x x Meprobamate x x Orphenadrine hydrochloride x x x x p-Benzoquinone x x x Phenol x x x x x Procainamide hydrochloride x x x Quinidine sulfate dehydrate x x x Resorcinol x x Rifampicin x x doi:10.14573/altex.1805181s2 ALTEX ##(#), SUPPLEMENTARY DATA 1 General Nervous Cardiovascular GI Chemical Liver Lung Blood Kidney cytotoxicity system system system Sodium Cyanate x x sodium oxalate x x x Sodium valproate x bx x x Thioridazine hydrochloride x x Valproic acid x x x x x GI: Gastrointestinal; CLP: Classification, labelling and packaging; NRU: Neutral Red Uptake; a Indirect effect: b chronic effect Tab. -

2019 Minnesota Chemicals of High Concern List
Minnesota Department of Health, Chemicals of High Concern List, 2019 Persistent, Bioaccumulative, Toxic (PBT) or very Persistent, very High Production CAS Bioaccumulative Use Example(s) and/or Volume (HPV) Number Chemical Name Health Endpoint(s) (vPvB) Source(s) Chemical Class Chemical1 Maine (CA Prop 65; IARC; IRIS; NTP Wood and textiles finishes, Cancer, Respiratory 11th ROC); WA Appen1; WA CHCC; disinfection, tissue 50-00-0 Formaldehyde x system, Eye irritant Minnesota HRV; Minnesota RAA preservative Gastrointestinal Minnesota HRL Contaminant 50-00-0 Formaldehyde (in water) system EU Category 1 Endocrine disruptor pesticide 50-29-3 DDT, technical, p,p'DDT Endocrine system Maine (CA Prop 65; IARC; IRIS; NTP PAH (chem-class) 11th ROC; OSPAR Chemicals of Concern; EuC Endocrine Disruptor Cancer, Endocrine Priority List; EPA Final PBT Rule for 50-32-8 Benzo(a)pyrene x x system TRI; EPA Priority PBT); Oregon P3 List; WA Appen1; Minnesota HRV WA Appen1; Minnesota HRL Dyes and diaminophenol mfg, wood preservation, 51-28-5 2,4-Dinitrophenol Eyes pesticide, pharmaceutical Maine (CA Prop 65; IARC; NTP 11th Preparation of amino resins, 51-79-6 Urethane (Ethyl carbamate) Cancer, Development ROC); WA Appen1 solubilizer, chemical intermediate Maine (CA Prop 65; IARC; IRIS; NTP Research; PAH (chem-class) 11th ROC; EPA Final PBT Rule for 53-70-3 Dibenzo(a,h)anthracene Cancer x TRI; WA PBT List; OSPAR Chemicals of Concern); WA Appen1; Oregon P3 List Maine (CA Prop 65; NTP 11th ROC); Research 53-96-3 2-Acetylaminofluorene Cancer WA Appen1 Maine (CA Prop 65; IARC; IRIS; NTP Lubricant, antioxidant, 55-18-5 N-Nitrosodiethylamine Cancer 11th ROC); WA Appen1 plastics stabilizer Maine (CA Prop 65; IRIS; NTP 11th Pesticide (EPA reg. -

June 16, 2014 Colleen Rogers Center for Drug Evaluation and Research
June 16, 2014 Colleen Rogers Center for Drug Evaluation and Research Food and Drug Administration Building 22, Room 5411 10903 New Hampshire Avenue Silver Spring, MD 20993 Re: Proposed Rule: Proposed Amendment of the Tentative Final Monograph, Federal Register, Vol. 78, No. 242, Tuesday, December 17, 2013. Docket identification (ID) number: FDA-1975-N-0012 Regulatory Information Number: 0910-AF69 Dear Ms. Rogers: The American Cleaning Institute (ACI)1 appreciates this opportunity to provide comments on the proposed rule to amend the 1994 tentative final monograph (the 1994 TFM) for over-the-counter (OTC) antiseptic drug products to establish conditions under which OTC consumer antiseptic products intended for use with water (referred to throughout as consumer antiseptic washes) are generally recognized as safe and effective. ACI has a specific interest in triclosan (TCS) within the proposed rule since our members produce consumer antiseptic wash products containing triclosan and manufacture triclosan. Triclosan-containing consumer antiseptic wash products play a beneficial role in the daily hygiene routines of millions of people throughout the U.S. and worldwide. They have been and are used safely and effectively in homes, hospitals, schools and workplaces every single day. Furthermore, triclosan and products containing it are regulated by a number of governmental bodies around the world and have a long track record of human and environmental safety which is supported by a multitude of science-based, transparent risk analyses. ACI members are concerned that FDA has not appropriately assessed the safety data that are available prior to proposing that additional safety data are necessary to support the safety of triclosan for this use. -

General Guidelines for NOI Forms B Through L and CWB NOI General Form (CWBNOI General.Pdf)
State of Hawaii Do NOT submit Department of Health Clean Water Branch this document. General Guidelines for NOI Forms B through L and CWB NOI General Form (CWBNOI_General.pdf) General Guidelines for Notice of Intent for Hawaii Administrative Rules, Chapter 11-55, Appendices B through L National Pollutant Discharge Elimination System (NPDES) Notice of General Permit Coverage (NGPC) For coverage under a specific NPDES General Permit, the following items are required to be submitted to the Clean Water Branch (CWB): A. CWB NOI General Form (CWBNOI_General.pdf) with Certifying Person’s original signature [via “Submit via Email” button and hard copy] B. General Permit Specific CWB NOI Form B, C, D, E, F, G, H, I, K, or L (CWBNOI_B.pdf through CWBNOI_L.doc) [via “Submit via Email” button, as applicable, and hard copy] C. All applicable attachments [via hard copy] D. $500 Filing Fee [Check made payable to “State of Hawaii”] E. Additional copies as required for Islands other than Oahu [see Notes V.D. and V.E. of the General Guidelines] TABLE OF CONTENTS Note Page General Information Applicable to All NOI Forms ......................................... 3 I. Introduction to the NPDES General Permit ........................................... 3 II. Class of Receiving State Waters Not Covered by NPDES General Permits .................. 4 III. Discharge Activities Covered by an NPDES General Permit .............................. 4 A. HAR, Chapter 11-55, Appendix B .............................................. 4 B. HAR, Chapter 11-55, Appendix C .............................................. 6 C. HAR, Chapter 11-55, Appendix D .............................................. 7 D. HAR, Chapter 11-55, Appendix E .............................................. 7 E. HAR, Chapter 11-55, Appendix F .............................................. 7 F. HAR, Chapter 11-55, Appendix G ............................................ -

State of Illinois Environmental Protection Agency Application for Environmental Laboratory Accreditation
State of Illinois Environmental Protection Agency Application for Environmental Laboratory Accreditation Attachment 6 Program: RCRA Field of Testing: Solid and Chemical Materials, Organic Matrix: Non Solid and Potable Chemical Accredited Water Materials Analyte: Status* Method (SW846): 1,2-Dibromo-3-chloropropane (DBCP) 8011 1,2-Dibromoethane (EDB) 8011 1,4-Dioxane 8015B 1-Butanol (n-Butyl alcohol) 8015B 1-Propanol 8015B 2-Butanone (Methyl ethyl ketone, MEK) 8015B 2-Chloroacrylonitrile 8015B 2-Methyl-1-propanol (Isobutyl alcohol) 8015B 2-Methylpyridine (2-Picoline) 8015B 2-Pentanone 8015B 2-Propanol (Isopropyl alcohol) 8015B 4-Methyl-2-pentanone (Methyl isobutyl ketone, 8015B MIBK) Acetone 8015B Acetonitrile 8015B Acrolein (Propenal) 8015B Acrylonitrile 8015B Allyl alcohol 8015B Crotonaldehyde 8015B Diesel range organics (DRO) 8015B Diethyl ether 8015B Ethanol 8015B Ethyl acetate 8015B Ethylene glycol 8015B Ethylene oxide 8015B Gasoline range organics (GRO) 8015B Hexafluoro-2-methyl-2-propanol 8015B * PI: Pending Initial Accreditation A: Accredited SP: Suspended WD: Accreditation Withdrawn Attachment 6: Page 1 of 62 Program: RCRA Field of Testing: Solid and Chemical Materials, Organic Matrix: Non Solid and Potable Chemical Accredited Water Materials Analyte: Status* Method (SW846): Hexafluoro-2-propanol 8015B Isopropyl benzene ((1-methylethyl) benzene) 8015B Methanol 8015B N-Nitrosodi-n-butylamine (N-Nitrosodibutylamine) 8015B o-Toluidine 8015B Paraldehyde 8015B Propionitrile (Ethyl cyanide) 8015B Pyridine 8015B t-Butyl alcohol 8015B