Tumor Necrosis Factor (TNF)-Receptor 1 and 2 Mediate Homeostatic Synaptic Plasticity of Denervated Mouse Dentate Granule Cells
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Rab3a As a Modulator of Homeostatic Synaptic Plasticity
Wright State University CORE Scholar Browse all Theses and Dissertations Theses and Dissertations 2014 Rab3A as a Modulator of Homeostatic Synaptic Plasticity Andrew G. Koesters Wright State University Follow this and additional works at: https://corescholar.libraries.wright.edu/etd_all Part of the Biomedical Engineering and Bioengineering Commons Repository Citation Koesters, Andrew G., "Rab3A as a Modulator of Homeostatic Synaptic Plasticity" (2014). Browse all Theses and Dissertations. 1246. https://corescholar.libraries.wright.edu/etd_all/1246 This Dissertation is brought to you for free and open access by the Theses and Dissertations at CORE Scholar. It has been accepted for inclusion in Browse all Theses and Dissertations by an authorized administrator of CORE Scholar. For more information, please contact [email protected]. RAB3A AS A MODULATOR OF HOMEOSTATIC SYNAPTIC PLASTICITY A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy By ANDREW G. KOESTERS B.A., Miami University, 2004 2014 Wright State University WRIGHT STATE UNIVERSITY GRADUATE SCHOOL August 22, 2014 I HEREBY RECOMMEND THAT THE DISSERTATION PREPARED UNDER MY SUPERVISION BY Andrew G. Koesters ENTITLED Rab3A as a Modulator of Homeostatic Synaptic Plasticity BE ACCEPTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF Doctor of Philosophy. Kathrin Engisch, Ph.D. Dissertation Director Mill W. Miller, Ph.D. Director, Biomedical Sciences Ph.D. Program Committee on Final Examination Robert E. W. Fyffe, Ph.D. Vice President for Research Dean of the Graduate School Mark Rich, M.D./Ph.D. David Ladle, Ph.D. F. Javier Alvarez-Leefmans, M.D./Ph.D. Lynn Hartzler, Ph.D. -

How-To Gnome-Look Guide
HHOOWW--TTOO Written by David D Lowe GGNNOOMMEE--LLOOOOKK GGUUIIDDEE hen I first joined the harddisk, say, ~/Pictures/Wallpapers. right-clicking on your desktop Ubuntu community, I and selecting the appropriate You may have noticed that gnome- button (you know which one!). Wwas extremely look.org separates wallpapers into impressed with the amount of different categories, according to the customization Ubuntu had to size of the wallpaper in pixels. For Don't let acronyms intimidate offer. People posted impressive the best quality, you want this to you; you don't have to know screenshots, and mentioned the match your screen resolution. If you what the letters stand for to themes they were using. They don't know what your screen know what it is. Basically, GTK is soon led me to gnome-look.org, resolution is, click System > the system GNOME uses to the number one place for GNOME Preferences > Screen Resolution. display things like buttons and visual customization. The However, Ubuntu stretches controls. GNOME is Ubuntu's screenshots there looked just as wallpapers quite nicely if you picked default desktop environment. I impressive, but I was very the wrong size, so you needn't fret will only be dealing with GNOME confused as to what the headings about it. on the sidebar meant, and I had customization here--sorry no idea how to use the files I SVG is a special image format that Kubuntu and Xubuntu folks! downloaded. Hopefully, this guide doesn't use pixels; it uses shapes Gnome-look.org distinguishes will help you learn what I found called vectors, which means you can between two versions of GTK: out the slow way. -
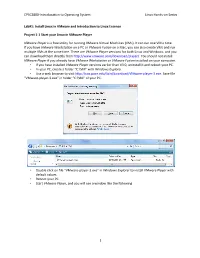
Install Linux in Vmware and Introduction to Linux Essence
CPSC2800‐Introducation to Operating System Linux Hands‐on Series Lab#1: Install Linux in VMware and Introduction to Linux Essence Project 1‐1 Start your Linux in VMware Player VMware Player is a free utility for running VMware Virtual Machines (VMs). It can run one VM a time. If you have VMware Workstation on a PC or VMware Fusion on a Mac, you can also create VMs and run multiple VMs at the same time. There are VMware Player versions for both Linux and Windows, and you can download them directly from http://www.vmware.com/ddownload/player/. You should not install VMware Player if you already have VMware Workstation or VMware Fusion installed on your computer. • If you have installed VMware Player versions earlier than V3.0, uninstall it and reboot your PC. • In your PC, create a folder “C:\VM” with Windows Expxplorer. • Use a web browser to visit http://csis.pace.edu/lixin/download/VVMware‐player‐3.exe. Save file “VMware‐player‐3.exe” in folder “C:\VM” of your PC. • Double click on file “VMware‐player‐3.exe” in Windoows Explorer to install VMware Player with default values. • Reboot your PC. • Start VMware Player, and you will see a window like the following. 1 CPSC2800‐Introducation to Operating System Linux Hands‐on Series • Click on menu item “File|Preferences…” 2 CPSC2800‐Introducation to Operating System Linux Hands‐on Series • In the “Preferences” window, uncheck for software updates, and click on the “Download Alll Components Now” button so you can later install VMware Tools in your new VMs without Internet access. -

Free As in Freedom
Daily Diet Free as in freedom ... • The freedom to run the program, for any purpose (freedom 0). Application Seen elsewhere Free Software Choices • The freedom to study how the program works, and adapt it to Text editor Wordpad Kate / Gedit/Vi/ Emacs your needs (freedom 1). Access to the source code is a precondition for this. Office Suite Microsoft Office KOffice / Open Office • The freedom to redistribute copies so you can help your Word Processor Microsoft Word Kword / Writer Presentation PowerPoint KPresenter / Impress neighbor (freedom 2). Spreadsheet Excel Kexl / Calc • The freedom to improve the program, and release your Mail & Info Manager Outlook Thunderbird / Evolution improvements to the public, so that the whole community benefits (freedom 3). Access to the source code is a Browser Safari, IE Konqueror / Firefox precondition for this. Chat client MSN, Yahoo, Gtalk, Kopete / Gaim IRC mIRC Xchat Non-Kernel parts = GNU (GNU is Not Unix) [gnu.org] Netmeeting Ekiga Kernel = Linux [kernel.org] PDF reader Acrobat Reader Kpdf / Xpdf/ Evince GNU Operating Syetem = GNU/Linux or GNU+Linux CD - burning Nero K3b / Gnome Toaster Distro – A flavor [distribution] of GNU/Linux os Music, video Winamp, Media XMMS, mplayer, xine, player rythmbox, totem Binaries ± Executable Terminal>shell>command line – interface to type in command Partition tool Partition Magic Gparted root – the superuser, administrator Graphics and Design Photoshop, GIMP, Image Magick & Corel Draw Karbon14,Skencil,MultiGIF The File system Animation Flash Splash Flash, f4l, Blender Complete list- linuxrsp.ru/win-lin-soft/table-eng.html, linuxeq.com/ Set up Broadband Ubuntu – set up- in terminal sudo pppoeconf. -

Technology User Guide Volume III: DRC INSIGHT
Technology User Guide Volume III: DRC INSIGHT WISCONSIN Data Recognition Corporation (DRC) 13490 Bass Lake Road Maple Grove, MN 55311 Wisconsin Service Line: 1-800-459-6530 DRC INSIGHT Portal: https://wi.drcedirect.com Email: [email protected] Revision Date: November 12, 2020 COPYRIGHT Copyright © 2020 Data Recognition Corporation The following items in DRC INSIGHT are protected by copyright law: • The User Guide. • All text and titles on the software’s entry and display, including the look and feel of the interaction of the windows, supporting menus, pop-up windows, and layout. DRC INSIGHT Online Learning System and DRC INSIGHT Portal are trademarked by Data Recognition Corporation. Any individuals or corporations who violate these copyrights and trademarks will be prosecuted under both criminal and civil laws, and any resulting products will be required to be withdrawn from the marketplace. The following are trademarks or registered trademarks of Microsoft Corporation in the United States and/or other countries: Internet Explorer Microsoft Windows Windows Vista Windows XP Windows 7 Windows 8 Windows 10 The following are trademarks or registered trademarks of Apple Corporation in the United States and/or other countries: Apple Macintosh Mac OS X and macOS iPad iPadOS iOS* *iOS is a trademark or registered trademark of Cisco in the U.S. and other countries and is used under license. Safari The following are trademarks or registered trademarks of Google Corporation in the United States and/or other countries. Chrome Chromebook Google Play The following is a trademark or registered trademark of Mozilla Corporation in the United States and/or other countries. -

Disruption of NMDA Receptor Function Prevents Normal Experience
Research Articles: Development/Plasticity/Repair Disruption of NMDA receptor function prevents normal experience-dependent homeostatic synaptic plasticity in mouse primary visual cortex https://doi.org/10.1523/JNEUROSCI.2117-18.2019 Cite as: J. Neurosci 2019; 10.1523/JNEUROSCI.2117-18.2019 Received: 16 August 2018 Revised: 7 August 2019 Accepted: 8 August 2019 This Early Release article has been peer-reviewed and accepted, but has not been through the composition and copyediting processes. The final version may differ slightly in style or formatting and will contain links to any extended data. Alerts: Sign up at www.jneurosci.org/alerts to receive customized email alerts when the fully formatted version of this article is published. Copyright © 2019 the authors Rodriguez et al. 1 Disruption of NMDA receptor function prevents normal experience-dependent homeostatic 2 synaptic plasticity in mouse primary visual cortex 3 4 Gabriela Rodriguez1,4, Lukas Mesik2,3, Ming Gao2,5, Samuel Parkins1, Rinki Saha2,6, 5 and Hey-Kyoung Lee1,2,3 6 7 8 1. Cell Molecular Developmental Biology and Biophysics (CMDB) Graduate Program, 9 Johns Hopkins University, Baltimore, MD 21218 10 2. Department of Neuroscience, Mind/Brain Institute, Johns Hopkins University, Baltimore, 11 MD 21218 12 3. Kavli Neuroscience Discovery Institute, Johns Hopkins University, Baltimore, MD 13 21218 14 4. Current address: Max Planck Florida Institute for Neuroscience, Jupiter, FL 33458 15 5. Current address: Division of Neurology, Barrow Neurological Institute, Pheonix, AZ 16 85013 17 6. Current address: Department of Psychiatry, Columbia University, New York, NY10032 18 19 Abbreviated title: NMDARs in homeostatic synaptic plasticity of V1 20 21 Corresponding Author: Hey-Kyoung Lee, Ph.D. -

Ubuntu® 1.4Inux Bible
Ubuntu® 1.4inux Bible William von Hagen 111c10,ITENNIAL. 18072 @WILEY 2007 •ICIOATENNIAl. Wiley Publishing, Inc. Acknowledgments xxi Introduction xxiii Part 1: Getting Started with Ubuntu Linux Chapter 1: The Ubuntu Linux Project 3 Background 4 Why Use Linux 4 What Is a Linux Distribution? 5 Introducing Ubuntu Linux 6 The Ubuntu Manifesto 7 Ubuntu Linux Release Schedule 8 Ubuntu Update and Maintenance Commitments 9 Ubuntu and the Debian Project 9 Why Choose Ubuntu? 10 Installation Requirements 11 Supported System Types 12 Hardware Requirements 12 Time Requirements 12 Ubuntu CDs 13 Support for Ubuntu Linux 14 Community Support and Information 14 Documentation 17 Commercial Support for Ubuntu Linux 18 Getting More Information About Ubuntu 19 Summary 20 Chapter 2: Installing Ubuntu 21 Getting a 64-bit or PPC Desktop CD 22 Booting the Desktop CD 22 Installing Ubuntu Linux from the Desktop CD 24 Booting Ubuntu Linux 33 Booting Ubuntu Linux an Dual-Boot Systems 33 The First Time You Boot Ubuntu Linux 34 Test-Driving Ubuntu Linux 34 Expioring the Desktop CD's Examples Folder 34 Accessing Your Hard Drive from the Desktop CD 36 Using Desktop CD Persistence 41 Copying Files to Other Machines Over a Network 43 Installing Windows Programs from the Desktop CD 43 Summary 45 ix Contents Chapter 3: Installing Ubuntu on Special-Purpose Systems 47 Overview of Dual-Boot Systems 48 Your Computer's Boot Process 48 Configuring a System for Dual-Booting 49 Repartitioning an Existing Disk 49 Getting a Different Install CD 58 Booting from a Server or Altemate -

LIFE Packages
LIFE packages Index Office automation Desktop Internet Server Web developpement Tele centers Emulation Health centers Graphics High Schools Utilities Teachers Multimedia Tertiary schools Programming Database Games Documentation Internet - Firefox - Browser - Epiphany - Nautilus - Ftp client - gFTP - Evolution - Mail client - Thunderbird - Internet messaging - Gaim - Gaim - IRC - XChat - Gaim - VoIP - Skype - Videomeeting - Gnome meeting - GnomeBittorent - P2P - aMule - Firefox - Download manager - d4x - Telnet - Telnet Web developpement - Quanta - Bluefish - HTML editor - Nvu - Any text editor - HTML galerie - Album - Web server - XAMPP - Collaborative publishing system - Spip Desktop - Gnome - Desktop - Kde - Xfce Graphics - Advanced image editor - The Gimp - KolourPaint - Simple image editor - gPaint - TuxPaint - CinePaint - Video editor - Kino - OpenOffice Draw - Vector vraphics editor - Inkscape - Dia - Diagram editor - Kivio - Electrical CAD - Electric - 3D modeller/render - Blender - CAD system - QCad Utilities - Calculator - gCalcTool - gEdit - gxEdit - Text editor - eMacs21 - Leafpad - Application finder - Xfce4-appfinder - Desktop search tool - Beagle - File explorer - Nautilus -Archive manager - File-Roller - Nautilus CD Burner - CD burner - K3B - GnomeBaker - Synaptic - System updates - apt-get - IPtables - Firewall - FireStarter - BackupPC - Backup - Amanda - gnome-terminal - Terminal - xTerm - xTerminal - Scanner - Xsane - Partition editor - gParted - Making image of disks - Partitimage - Mirroring over network - UDP Cast -

Homeostatic Synaptic Plasticity: Local and Global Mechanisms for Stabilizing Neuronal Function
Downloaded from http://cshperspectives.cshlp.org/ on September 25, 2021 - Published by Cold Spring Harbor Laboratory Press Homeostatic Synaptic Plasticity: Local and Global Mechanisms for Stabilizing Neuronal Function Gina Turrigiano Department of Biology and Center for Behavioral Genomics, Brandeis University, Waltham, Massachusetts 02493 Correspondence: [email protected] Neural circuits must maintain stable function in the face of many plastic challenges, includ- ing changes in synapse number and strength, during learning and development. Recent work has shown that these destabilizing influences are counterbalanced by homeostatic plasticity mechanisms that act to stabilize neuronal and circuit activity. One such mechanism is syn- aptic scaling, which allows neurons to detect changes in their own firing rates through a set of calcium-dependent sensors that then regulate receptor trafficking to increase or decrease the accumulation of glutamate receptors at synaptic sites. Additional homeostatic mechanisms may allow local changes in synaptic activation to generate local synaptic adaptations, and network-wide changes in activity to generate network-wide adjustments in the balance between excitation and inhibition. The signaling pathways underlying these various forms of homeostatic plasticity are currently under intense scrutiny, and although dozens of mol- ecular pathways have now been implicated in homeostatic plasticity, a clear picture of how homeostatic feedback is structured at the molecular level has not yet emerged. On a functional level, neuronal networks likely use this complex set of regulatory mechanisms to achieve homeostasis over a wide range of temporal and spatial scales. ore than 50 years ago, Walter Cannon eter that is subject to homeostatic regulation. Mmarveled that “somehow the unstable During development billions of neurons wire stuff of which we are composed has learned themselves up into complex networks and man- the trick of maintaining stability” (Cannon age to reach a state where they can generate— 1932). -

Install Gnome Software Center Arch
Install gnome software center arch Upstream URL: License(s): GPL2. Maintainers: Jan Steffens. Package Size: MB. Installed Size: Installed Size: MB. gnome-software will be available as a preview in It can install, remove applications on systems with PackageKit. It can install updates on Gnome software will not start / Applications & Desktop. A quick video on Gnome Software Center in Arch Linux. Gnome unstable repository. There is a component called Polkit that is used by many applications to request root permissions to do things (it can do so because it's a. GNOME Software on #archlinux with native PackageKit backend, and this is a gui for installing software, ala ubuntu software manager, but distro This is some kind of Ubuntu Software Centre, with comments and all that. Need help installing Gnome Software Center for Arch Linux? Here are some instructions: Click DOWNLOAD HERE in the menu. Download the file. Make the file. I had to install it with along with packagekit. This is what's missing to make Antergos *the* beginner-friendly Arch-based distro, or general So, it is not a bad idea for the “Gnome Software Center” to include by default. GNOME software software center graphic that we will find the default in future releases of Fedora in addition to being installed in Arch Linux Please help me to install GNOME Software on. GNOME Software Will Work On Arch Linux With PackageKit the Alpm/Pacman back-end for using this GNOME application to install and. From: Sriram Ramkrishna ; To: desktop-devel-list devel-list gnome org>; Subject: gnome- software/packagekit. -

ANJUTA – Um Breve Tutorial 1
ANJUTA – Um breve tutorial 1 Sumário Anjuta um breve tutorial..................................................................................................................2 Definição.....................................................................................................................................2 Recursos do Anjuta.....................................................................................................................3 Antes de começar........................................................................................................................4 O primeiro projeto...........................................................................................................................5 Incrementando o projeto............................................................................................................12 Invocando os poderes do Glade................................................................................................12 O esqueleto básico do programa...............................................................................................14 Qual comando usar para compilar?...........................................................................................16 Compilando e rodando o projeto...................................................................................................17 Dividir para conquistar!............................................................................................................17 Criando os novos arquivos...................................................................................................18 -
Hello Everyone. I Introduce You Voyager GE 19.04 That Continues the Adventure with the Desktop Gnome Shell Version 3.32 by Introducing New Features
Hello everyone. I introduce you Voyager GE 19.04 that continues the adventure with the desktop Gnome Shell version 3.32 by introducing new features. With the promise finally realized to have a light Gnome system, fast, fluid and powerful. This version is based on the Linux 5.0 kernel and distribution Ubuntu Disco Dingo . 19.04 is an intermediate version upgrade nine months preparing the future version 20.04 LTS - Long-term Support - 5 years that happens every two years, when Voyager will be available in 2 versions, GNOME Shell and Xfce. Also you can choose according to your wishes and capacity machines. The general idea of Voyager is to introduce in Gnome Gnome Shell preinstalled extensions and scripts grouped in a box that optimize the system with a choice of necessary software. A video presentation and pictures are available. Soon for this new human and digital adventure. Voyager wants multi-profile and multi-task in an environment aesthetic and immersive as possible and that, from the origins of traveling, so that time spent on your machine is the most pleasant. In summary, the general idea is that for each profile, we have options available types that can activate or not. Voyager GE 19.04 based on Ubuntu 19.04 will cycle nine months of support and updates. For information, these are intermediate versions that prepare Future LTS. You can then seamlessly transition to 19.10 with automatic up-to-date system and so on until the next LTS. Voyager GE is not a distribution with its deposits but a customized variant of Ubuntu as mentioned above, with all its official repositories.