Lysosomal Membrane Transport Physiological
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genes in Eyecare Geneseyedoc 3 W.M
Genes in Eyecare geneseyedoc 3 W.M. Lyle and T.D. Williams 15 Mar 04 This information has been gathered from several sources; however, the principal source is V. A. McKusick’s Mendelian Inheritance in Man on CD-ROM. Baltimore, Johns Hopkins University Press, 1998. Other sources include McKusick’s, Mendelian Inheritance in Man. Catalogs of Human Genes and Genetic Disorders. Baltimore. Johns Hopkins University Press 1998 (12th edition). http://www.ncbi.nlm.nih.gov/Omim See also S.P.Daiger, L.S. Sullivan, and B.J.F. Rossiter Ret Net http://www.sph.uth.tmc.edu/Retnet disease.htm/. Also E.I. Traboulsi’s, Genetic Diseases of the Eye, New York, Oxford University Press, 1998. And Genetics in Primary Eyecare and Clinical Medicine by M.R. Seashore and R.S.Wappner, Appleton and Lange 1996. M. Ridley’s book Genome published in 2000 by Perennial provides additional information. Ridley estimates that we have 60,000 to 80,000 genes. See also R.M. Henig’s book The Monk in the Garden: The Lost and Found Genius of Gregor Mendel, published by Houghton Mifflin in 2001 which tells about the Father of Genetics. The 3rd edition of F. H. Roy’s book Ocular Syndromes and Systemic Diseases published by Lippincott Williams & Wilkins in 2002 facilitates differential diagnosis. Additional information is provided in D. Pavan-Langston’s Manual of Ocular Diagnosis and Therapy (5th edition) published by Lippincott Williams & Wilkins in 2002. M.A. Foote wrote Basic Human Genetics for Medical Writers in the AMWA Journal 2002;17:7-17. A compilation such as this might suggest that one gene = one disease. -

Sialic Acid Storage Disease
Sialic acid storage disease Description Sialic acid storage disease is an inherited disorder that primarily affects the nervous system. People with sialic acid storage disease have signs and symptoms that may vary widely in severity. This disorder is generally classified into one of three forms: infantile free sialic acid storage disease, Salla disease, and intermediate severe Salla disease. Infantile free sialic acid storage disease (ISSD) is the most severe form of this disorder. Babies with this condition have severe developmental delay, weak muscle tone ( hypotonia), and failure to gain weight and grow at the expected rate (failure to thrive). They may have unusual facial features that are often described as "coarse," seizures, bone malformations, an enlarged liver and spleen (hepatosplenomegaly), and an enlarged heart (cardiomegaly). The abdomen may be swollen due to the enlarged organs and an abnormal buildup of fluid in the abdominal cavity (ascites). Affected infants may have a condition called hydrops fetalis in which excess fluid accumulates in the body before birth. Children with this severe form of the condition usually live only into early childhood. Salla disease is a less severe form of sialic acid storage disease. Babies with Salla disease usually begin exhibiting hypotonia during the first year of life and go on to experience progressive neurological problems. Signs and symptoms of Salla disease include intellectual disability and developmental delay, seizures, problems with movement and balance (ataxia), abnormal tensing of the muscles (spasticity), and involuntary slow, sinuous movements of the limbs (athetosis). Individuals with Salla disease usually survive into adulthood. People with intermediate severe Salla disease have signs and symptoms that fall between those of ISSD and Salla disease in severity. -

Centometabolic® COMBINING GENETIC and BIOCHEMICAL TESTING for the FAST and COMPREHENSIVE DIAGNOSTIC of METABOLIC DISORDERS
CentoMetabolic® COMBINING GENETIC AND BIOCHEMICAL TESTING FOR THE FAST AND COMPREHENSIVE DIAGNOSTIC OF METABOLIC DISORDERS GENE ASSOCIATED DISEASE(S) ABCA1 HDL deficiency, familial, 1; Tangier disease ABCB4 Cholestasis, progressive familial intrahepatic 3 ABCC2 Dubin-Johnson syndrome ABCD1 Adrenoleukodystrophy; Adrenomyeloneuropathy, adult ABCD4 Methylmalonic aciduria and homocystinuria, cblJ type ABCG5 Sitosterolemia 2 ABCG8 Sitosterolemia 1 ACAT1 Alpha-methylacetoacetic aciduria ADA Adenosine deaminase deficiency AGA Aspartylglucosaminuria AGL Glycogen storage disease IIIa; Glycogen storage disease IIIb AGPS Rhizomelic chondrodysplasia punctata, type 3 AGXT Hyperoxaluria, primary, type 1 ALAD Porphyria, acute hepatic ALAS2 Anemia, sideroblastic, 1; Protoporphyria, erythropoietic, X-linked ALDH4A1 Hyperprolinemia, type II ALDOA Glycogen storage disease XII ALDOB Fructose intolerance, hereditary ALG3 Congenital disorder of glycosylation, type Id ALPL Hypophosphatasia, adult; Hypophosphatasia, childhood; Hypophosphatasia, infantile; Odontohypophosphatasia ANTXR2 Hyaline fibromatosis syndrome APOA2 Hypercholesterolemia, familial, modifier of APOA5 Hyperchylomicronemia, late-onset APOB Hypercholesterolemia, familial, 2 APOC2 Hyperlipoproteinemia, type Ib APOE Sea-blue histiocyte disease; Hyperlipoproteinemia, type III ARG1 Argininemia ARSA Metachromatic leukodystrophy ARSB Mucopolysaccharidosis type VI (Maroteaux-Lamy) 1 GENE ASSOCIATED DISEASE(S) ASAH1 Farber lipogranulomatosis; Spinal muscular atrophy with progressive myoclonic epilepsy -

Table S1. Disease Classification and Disease-Reaction Association
Table S1. Disease classification and disease-reaction association Disorder class Associated reactions cross Disease Ref[Goh check et al. -

Antenatal Diagnosis of Inborn Errors Ofmetabolism
816 ArchivesofDiseaseinChildhood 1991;66: 816-822 CURRENT PRACTICE Arch Dis Child: first published as 10.1136/adc.66.7_Spec_No.816 on 1 July 1991. Downloaded from Antenatal diagnosis of inborn errors of metabolism M A Cleary, J E Wraith The introduction of experimental treatment for Sample requirement and techniques used in lysosomal storage disorders and the increasing prenatal diagnosis understanding of the molecular defects behind By far the majority of antenatal diagnoses are many inborn errors have overshadowed the fact performed on samples obtained by either that for many affected families the best that can amniocentesis or chorion villus biopsy. For be offered is a rapid, accurate prenatal diag- some disorders, however, the defect is not nostic service. Many conditions remain at best detectable in this material and more invasive only partially treatable and as a consequence the methods have been applied to obtain a diagnos- majority of parents seek antenatal diagnosis in tic sample. subsequent pregnancies, particularly for those disorders resulting in a poor prognosis in terms of either life expectancy or normal neurological FETAL LIVER BIOPSY development. Fetal liver biopsy has been performed to The majority of inborn errors result from a diagnose ornithine carbamoyl transferase defi- specific enzyme deficiency, but in some the ciency and primary hyperoxaluria type 1. primary defect is in a transport system or Glucose-6-phosphatase deficiency (glycogen enzyme cofactor. In some conditions the storage disease type I) could also be detected by biochemical defect is limited to specific tissues this method. The technique, however, is inva- only and this serves to restrict the material avail- sive and can be performed by only a few highly able for antenatal diagnosis for these disorders. -

Transporters
Alexander, S. P. H., Kelly, E., Mathie, A., Peters, J. A., Veale, E. L., Armstrong, J. F., Faccenda, E., Harding, S. D., Pawson, A. J., Sharman, J. L., Southan, C., Davies, J. A., & CGTP Collaborators (2019). The Concise Guide to Pharmacology 2019/20: Transporters. British Journal of Pharmacology, 176(S1), S397-S493. https://doi.org/10.1111/bph.14753 Publisher's PDF, also known as Version of record License (if available): CC BY Link to published version (if available): 10.1111/bph.14753 Link to publication record in Explore Bristol Research PDF-document This is the final published version of the article (version of record). It first appeared online via Wiley at https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14753. Please refer to any applicable terms of use of the publisher. University of Bristol - Explore Bristol Research General rights This document is made available in accordance with publisher policies. Please cite only the published version using the reference above. Full terms of use are available: http://www.bristol.ac.uk/red/research-policy/pure/user-guides/ebr-terms/ S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2019/20: Transporters. British Journal of Pharmacology (2019) 176, S397–S493 THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Transporters Stephen PH Alexander1 , Eamonn Kelly2, Alistair Mathie3 ,JohnAPeters4 , Emma L Veale3 , Jane F Armstrong5 , Elena Faccenda5 ,SimonDHarding5 ,AdamJPawson5 , Joanna L Sharman5 , Christopher Southan5 , Jamie A Davies5 and CGTP Collaborators 1School of Life Sciences, -

Metabolic Evaluation of Epilepsy: a Diagnostic Algorithm with Focus on Treatable Conditions
Zurich Open Repository and Archive University of Zurich Main Library Strickhofstrasse 39 CH-8057 Zurich www.zora.uzh.ch Year: 2018 Metabolic Evaluation of Epilepsy: A Diagnostic Algorithm With Focus on Treatable Conditions van Karnebeek, Clara D M ; Sayson, Bryan ; Lee, Jessica J Y ; Tseng, Laura A ; Blau, Nenad ; Horvath, Gabriella A ; Ferreira, Carlos R Abstract: Although inborn errors of metabolism do not represent the most common cause of seizures, their early identification is of utmost importance, since many will require therapeutic measures beyond that of common anti-epileptic drugs, either in order to control seizures, or to decrease the risk of neurode- generation. We translate the currently-known literature on metabolic etiologies of epilepsy (268 inborn errors of metabolism belonging to 21 categories, with 74 treatable errors), into a 2-tiered diagnostic algo- rithm, with the first-tier comprising accessible, affordable, and less invasive screening tests in urineand blood, with the potential to identify the majority of treatable conditions, while the second-tier tests are ordered based on individual clinical signs and symptoms. This resource aims to support the pediatri- cian, neurologist, biochemical, and clinical geneticists in early identification of treatable inborn errors of metabolism in a child with seizures, allowing for timely initiation of targeted therapy with the potential to improve outcomes. DOI: https://doi.org/10.3389/fneur.2018.01016 Posted at the Zurich Open Repository and Archive, University of Zurich ZORA URL: https://doi.org/10.5167/uzh-161470 Journal Article Published Version The following work is licensed under a Creative Commons: Attribution 4.0 International (CC BY 4.0) License. -
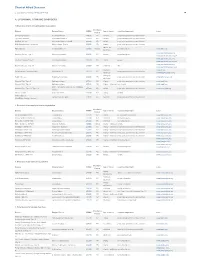
Pdf NTSAD Chart of Allied Diseases
Chart of Allied Diseases Last Updated: Monday, 19 May 2014 17:02 A. LYSOSOMAL STORAGE DISORDERS 1) Disorders of lipid and sphingolipid degradation Inheritance Disease Enzyme Defect OMIM# Age of Onset Cognitive Impairment Links Pattern GM1 Gangliosidosis b-Galactosidase-1 230500 AR variable progressive psychomotor deterioration Tay-Sachs Disease b-Hexosaminidase A 272800 AR variable progressive psychomotor deterioration Sandhoff Disease b-Hexosaminidases A and B 268800 AR variable progressive psychomotor deterioration GM2 Gangliodisosis, AB variant GM2 Activator Protein 272750 AR infancy progressive psychomotor deterioration adolesence - Fabry Disease 8-Galactosidase A 301500 X-linked normal intelligence www.fabry.org adulthood www.gaucherdisease.org, Gaucher Disease, Type 1 Glucocerebrosidase 230800 AR variable normal intelligence www.gaucherdisease.org.uk www.gaucherdisease.org, Gaucher Disease, Type II Glucocerebrosidase 230900 AR infancy severe www.gaucherdisease.org.uk www.gaucherdisease.org, Gaucher Disease, Type III Glucocerebrosidase 231000 AR childhood mild www.gaucherdisease.org.uk infancy to www.ulf.org, Metachromatic Leukodystrophy Arylsulfatase A 250100 AR progressive psychomotor deterioration adulthood www.MLDFoundation.org infancy to Krabbe Disease Galactosylceramidase 245200 AR progressive psychomotor deterioration www.huntershope.org adulthood Niemann-Pick, Type A Sphingomyelinase 257200 AR infancy progressive psychomotor deterioration www.nnpdf.org Niemann-Pick, Type B Sphingomyelinase 607616 AR infancy - childhood none -

Whole Exome Sequencing Gene Package Skeletal Dysplasia, Version 2.1, 31-1-2020
Whole Exome Sequencing Gene package Skeletal Dysplasia, Version 2.1, 31-1-2020 Technical information DNA was enriched using Agilent SureSelect DNA + SureSelect OneSeq 300kb CNV Backbone + Human All Exon V7 capture and paired-end sequenced on the Illumina platform (outsourced). The aim is to obtain 10 Giga base pairs per exome with a mapped fraction of 0.99. The average coverage of the exome is ~50x. Duplicate and non-unique reads are excluded. Data are demultiplexed with bcl2fastq Conversion Software from Illumina. Reads are mapped to the genome using the BWA-MEM algorithm (reference: http://bio-bwa.sourceforge.net/). Variant detection is performed by the Genome Analysis Toolkit HaplotypeCaller (reference: http://www.broadinstitute.org/gatk/). The detected variants are filtered and annotated with Cartagenia software and classified with Alamut Visual. It is not excluded that pathogenic mutations are being missed using this technology. At this moment, there is not enough information about the sensitivity of this technique with respect to the detection of deletions and duplications of more than 5 nucleotides and of somatic mosaic mutations (all types of sequence changes). HGNC approved Phenotype description including OMIM phenotype ID(s) OMIM median depth % covered % covered % covered gene symbol gene ID >10x >20x >30x ABCC9 Atrial fibrillation, familial, 12, 614050 601439 65 100 100 95 Cardiomyopathy, dilated, 1O, 608569 Hypertrichotic osteochondrodysplasia, 239850 ACAN Short stature and advanced bone age, with or without early-onset osteoarthritis -

The Myriad Foresight® Carrier Screen
The Myriad Foresight® Carrier Screen 180 Kimball Way | South San Francisco, CA 94080 www.myriadwomenshealth.com | [email protected] | (888) 268-6795 The Myriad Foresight® Carrier Screen - Disease Reference Book 11-beta-hydroxylase-deficient Congenital Adrenal Hyperplasia ...............................................................................................................................................................................8 6-pyruvoyl-tetrahydropterin Synthase Deficiency....................................................................................................................................................................................................10 ABCC8-related Familial Hyperinsulinism..................................................................................................................................................................................................................12 Adenosine Deaminase Deficiency ............................................................................................................................................................................................................................14 Alpha Thalassemia ....................................................................................................................................................................................................................................................16 Alpha-mannosidosis ..................................................................................................................................................................................................................................................18 -

Download CGT Exome V2.0
CGT Exome version 2. -

SSIEM Classification of Inborn Errors of Metabolism 2011
SSIEM classification of Inborn Errors of Metabolism 2011 Disease group / disease ICD10 OMIM 1. Disorders of amino acid and peptide metabolism 1.1. Urea cycle disorders and inherited hyperammonaemias 1.1.1. Carbamoylphosphate synthetase I deficiency 237300 1.1.2. N-Acetylglutamate synthetase deficiency 237310 1.1.3. Ornithine transcarbamylase deficiency 311250 S Ornithine carbamoyltransferase deficiency 1.1.4. Citrullinaemia type1 215700 S Argininosuccinate synthetase deficiency 1.1.5. Argininosuccinic aciduria 207900 S Argininosuccinate lyase deficiency 1.1.6. Argininaemia 207800 S Arginase I deficiency 1.1.7. HHH syndrome 238970 S Hyperammonaemia-hyperornithinaemia-homocitrullinuria syndrome S Mitochondrial ornithine transporter (ORNT1) deficiency 1.1.8. Citrullinemia Type 2 603859 S Aspartate glutamate carrier deficiency ( SLC25A13) S Citrin deficiency 1.1.9. Hyperinsulinemic hypoglycemia and hyperammonemia caused by 138130 activating mutations in the GLUD1 gene 1.1.10. Other disorders of the urea cycle 238970 1.1.11. Unspecified hyperammonaemia 238970 1.2. Organic acidurias 1.2.1. Glutaric aciduria 1.2.1.1. Glutaric aciduria type I 231670 S Glutaryl-CoA dehydrogenase deficiency 1.2.1.2. Glutaric aciduria type III 231690 1.2.2. Propionic aciduria E711 232000 S Propionyl-CoA-Carboxylase deficiency 1.2.3. Methylmalonic aciduria E711 251000 1.2.3.1. Methylmalonyl-CoA mutase deficiency 1.2.3.2. Methylmalonyl-CoA epimerase deficiency 251120 1.2.3.3. Methylmalonic aciduria, unspecified 1.2.4. Isovaleric aciduria E711 243500 S Isovaleryl-CoA dehydrogenase deficiency 1.2.5. Methylcrotonylglycinuria E744 210200 S Methylcrotonyl-CoA carboxylase deficiency 1.2.6. Methylglutaconic aciduria E712 250950 1.2.6.1. Methylglutaconic aciduria type I E712 250950 S 3-Methylglutaconyl-CoA hydratase deficiency 1.2.6.2.