The Cytolethal Distending Toxin from the Chancroid Bacterium Haemophilus Ducreyi Induces Cell-Cycle Arrest in the G2 Phase
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Parinaud's Oculoglandular Syndrome
Tropical Medicine and Infectious Disease Case Report Parinaud’s Oculoglandular Syndrome: A Case in an Adult with Flea-Borne Typhus and a Review M. Kevin Dixon 1, Christopher L. Dayton 2 and Gregory M. Anstead 3,4,* 1 Baylor Scott & White Clinic, 800 Scott & White Drive, College Station, TX 77845, USA; [email protected] 2 Division of Critical Care, Department of Medicine, University of Texas Health, San Antonio, 7703 Floyd Curl Drive, San Antonio, TX 78229, USA; [email protected] 3 Medical Service, South Texas Veterans Health Care System, San Antonio, TX 78229, USA 4 Division of Infectious Diseases, Department of Medicine, University of Texas Health, San Antonio, 7703 Floyd Curl Drive, San Antonio, TX 78229, USA * Correspondence: [email protected]; Tel.: +1-210-567-4666; Fax: +1-210-567-4670 Received: 7 June 2020; Accepted: 24 July 2020; Published: 29 July 2020 Abstract: Parinaud’s oculoglandular syndrome (POGS) is defined as unilateral granulomatous conjunctivitis and facial lymphadenopathy. The aims of the current study are to describe a case of POGS with uveitis due to flea-borne typhus (FBT) and to present a diagnostic and therapeutic approach to POGS. The patient, a 38-year old man, presented with persistent unilateral eye pain, fever, rash, preauricular and submandibular lymphadenopathy, and laboratory findings of FBT: hyponatremia, elevated transaminase and lactate dehydrogenase levels, thrombocytopenia, and hypoalbuminemia. His condition rapidly improved after starting doxycycline. Soon after hospitalization, he was diagnosed with uveitis, which responded to topical prednisolone. To derive a diagnostic and empiric therapeutic approach to POGS, we reviewed the cases of POGS from its various causes since 1976 to discern epidemiologic clues and determine successful diagnostic techniques and therapies; we found multiple cases due to cat scratch disease (CSD; due to Bartonella henselae) (twelve), tularemia (ten), sporotrichosis (three), Rickettsia conorii (three), R. -

Hemolysin from Escherichia Coli Uses Endogenous Amplification Through P2X Receptor Activation to Induce Hemolysis
␣-Hemolysin from Escherichia coli uses endogenous amplification through P2X receptor activation to induce hemolysis Marianne Skalsa, Niklas R. Jorgensenb, Jens Leipzigera, and Helle A. Praetoriusa,1 aDepartment of Physiology and Biophysics, Water and Salt Research Center, Aarhus University, Ole Worms Alle 1160, 8000 Aarhus C, Denmark; and bDepartment for Clinical Biochemistry, Roskilde Hospital, Koegevej 3-7, 4000 Roskilde, Denmark Edited by Sucharit Bhakdi, University of Mainz, Mainz, Germany, and accepted by the Editorial Board January 6, 2009 (received for review July 22, 2008) Escherichia coli is the dominant facultative bacterium in the normal and are referred to as P2X1–7. All P2X receptors are permeable to intestinal flora. E. coli is, however, also responsible for the majority small monovalent cations and some have significant calcium per- of serious extraintestinal infections. There are distinct serotypical meability (11). Here we show that human, murine, and equine differences between facultative and invasive E. coli strains. Inva- erythrocytes use a combination of P2X1 and P2X7 receptor acti- sive strains frequently produce virulence factors such as ␣-hemolysin vation for full HlyA-induced hemolysis to occur. This is particularly (HlyA), which causes hemolysis by forming pores in the erythrocyte interesting, as prolonged stimulation of P2X7 receptors are known membrane. The present study reveals that this pore formation to increase the plasma membrane permeability to an extent that triggers purinergic receptor activation to mediate the full hemo- eventually leads to lysis of certain cells (12). In macrophages it has lytic action. Non-selective ATP-receptor (P2) antagonists (PPADS, been shown that pannexin1, a recently discovered pore-forming suramin) and ATP scavengers (apyrase, hexokinase) concentration protein, is required for this increment in permeability (12, 13). -

Reportable Diseases and Conditions
KINGS COUNTY DEPARTMENT of PUBLIC HEALTH 330 CAMPUS DRIVE, HANFORD, CA 93230 REPORTABLE DISEASES AND CONDITIONS Title 17, California Code of Regulations, §2500, requires that known or suspected cases of any of the diseases or conditions listed below are to be reported to the local health jurisdiction within the specified time frame: REPORT IMMEDIATELY BY PHONE During Business Hours: (559) 852-2579 After Hours: (559) 852-2720 for Immediate Reportable Disease and Conditions Anthrax Escherichia coli: Shiga Toxin producing (STEC), Rabies (Specify Human or Animal) Botulism (Specify Infant, Foodborne, Wound, Other) including E. coli O157:H7 Scrombroid Fish Poisoning Brucellosis, Human Flavivirus Infection of Undetermined Species Shiga Toxin (Detected in Feces) Cholera Foodborne Disease (2 or More Cases) Smallpox (Variola) Ciguatera Fish Poisoning Hemolytic Uremic Syndrome Tularemia, human Dengue Virus Infection Influenza, Novel Strains, Human Viral Hemorrhagic Fever (Crimean-Congo, Ebola, Diphtheria Measles (Rubeola) Lassa, and Marburg Viruses) Domonic Acid Poisoning (Amnesic Shellfish Meningococcal Infections Yellow Fever Poisoning) Novel Virus Infection with Pandemic Potential Zika Virus Infection Paralytic Shellfish Poisoning Plague (Specify Human or Animal) Immediately report the occurrence of any unusual disease OR outbreaks of any disease. REPORT BY PHONE, FAX, MAIL WITHIN ONE (1) WORKING DAY Phone: (559) 852-2579 Fax: (559) 589-0482 Mail: 330 Campus Drive, Hanford 93230 Conditions may also be reported electronically via the California -

Disposal of Toxin Heptamers by Extracellular Vesicle Formation and Lysosomal Degradation
toxins Article Major Determinants of Airway Epithelial Cell Sensitivity to S. aureus Alpha-Toxin: Disposal of Toxin Heptamers by Extracellular Vesicle Formation and Lysosomal Degradation Nils Möller 1,* , Sabine Ziesemer 1, Christian Hentschker 2, Uwe Völker 2 and Jan-Peter Hildebrandt 1 1 Animal Physiology and Biochemistry, University of Greifswald, Felix Hausdorff-Strasse 1, D-17489 Greifswald, Germany; [email protected] (S.Z.); [email protected] (J.-P.H.) 2 Department of Functional Genomics, Interfaculty Institute for Genetics and Functional Genomics, University Medicine Greifswald, Felix Hausdorff-Strasse 8, D-17475 Greifswald, Germany; [email protected] (C.H.); [email protected] (U.V.) * Correspondence: [email protected] Abstract: Alpha-toxin is a major virulence factor of Staphylococcus aureus. Monomer binding to host cell membranes results in the formation of heptameric transmembrane pores. Among human model airway epithelial cell lines, A549 cells were most sensitive toward the toxin followed by 16HBE14o- and S9 cells. In this study we investigated the processes of internalization of pore-containing plasma membrane areas as well as potential pathways for heptamer degradation (lysosomal, proteasomal) or disposal (formation of exosomes/micro-vesicles). The abundance of toxin heptamers upon applying an alpha-toxin pulse to the cells declined both in extracts of whole cells and of cellular membranes of Citation: Möller, N.; Ziesemer, S.; S9 cells, but not in those of 16HBE14o- or A549 cells. Comparisons of heptamer degradation rates un- Hentschker, C.; Völker, U.; der inhibition of lysosomal or proteasomal degradation revealed that an important route of heptamer Hildebrandt, J.-P. -

2012 Case Definitions Infectious Disease
Arizona Department of Health Services Case Definitions for Reportable Communicable Morbidities 2012 TABLE OF CONTENTS Definition of Terms Used in Case Classification .......................................................................................................... 6 Definition of Bi-national Case ............................................................................................................................................. 7 ------------------------------------------------------------------------------------------------------- ............................................... 7 AMEBIASIS ............................................................................................................................................................................. 8 ANTHRAX (β) ......................................................................................................................................................................... 9 ASEPTIC MENINGITIS (viral) ......................................................................................................................................... 11 BASIDIOBOLOMYCOSIS ................................................................................................................................................. 12 BOTULISM, FOODBORNE (β) ....................................................................................................................................... 13 BOTULISM, INFANT (β) ................................................................................................................................................... -

Transport Proteins Promoting Escherichia Coli Pathogenesis
Microbial Pathogenesis 71-72 (2014) 41e55 Contents lists available at ScienceDirect Microbial Pathogenesis journal homepage: www.elsevier.com/locate/micpath Transport proteins promoting Escherichia coli pathogenesis Fengyi Tang 1, Milton H. Saier Jr. * Department of Molecular Biology, Division of Biological Sciences, University of California at San Diego, La Jolla, CA 92093-0116, USA article info abstract Article history: Escherichia coli is a genetically diverse species infecting hundreds of millions of people worldwide Received 26 November 2013 annually. We examined seven well-characterized E. coli pathogens causing urinary tract infections, Received in revised form gastroenteritis, pyelonephritis and haemorrhagic colitis. Their transport proteins were identified and 19 March 2014 compared with each other and a non-pathogenic E. coli K12 strain to identify transport proteins related Accepted 20 March 2014 to pathogenesis. Each pathogen possesses a unique set of protein secretion systems for export to the cell Available online 18 April 2014 surface or for injecting effector proteins into host cells. Pathogens have increased numbers of iron siderophore receptors and ABC iron uptake transporters, but the numbers and types of low-affinity Keywords: Escherichia coli secondary iron carriers were uniform in all strains. The presence of outer membrane iron complex re- fi Pathogenesis ceptors and high-af nity ABC iron uptake systems correlated, suggesting co-evolution. Each pathovar Transporters encodes a different set of pore-forming toxins and virulence-related outer membrane proteins lacking in Toxins K12. Intracellular pathogens proved to have a characteristically distinctive set of nutrient uptake porters, Iron acquisition different from those of extracellular pathogens. The results presented in this report provide information Intra vs. -

Sexually Transmitted Diseases/Infections Chancroid
Sexually Transmitted Diseases/Infections Chancroid Chancroid is a bacterial infection caused by Haemophilus ducreyi. It is spread by sexual contact and results in genital ulcers. Chancroid is a reportable genital ulcer condition that is rarely seen in North Carolina. When infection does occur, it is usually associated with sporadic outbreaks. Chancroid, as well as genital herpes and syphilis, is a risk factor in the transmission of HIV infection. Chancroid lesions may be difficult to distinguish from ulcers caused by genital herpes or syphilis. A physician must therefore diagnose the infection by excluding other diseases with similar symptoms. The combination of a painful genital ulcer and tender suppurative inguinal adenopathy suggests the diagnosis of chancroid. A probable diagnosis of chancroid, for both clinical and surveillance purposes, can be made if all of the following criteria are met: 1) the patient has one or more painful genital ulcers; 2) the patient has no evidence of T. pallidum infection by darkfield examination of ulcer exudate or by a serologic test for syphilis performed at least 7 days after onset of ulcers; 3) the clinical presentation, appearance of genital ulcers and, if present, regional lymphadenopathy are typical for chancroid; and 4) a test for HSV performed on the ulcer exudate is negative. A definitive diagnosis of chancroid requires the identification of H. ducreyi on special culture media that is not widely available from commercial sources; even when these media are used, sensitivity is less than 80 percent. No FDA- cleared PCR test for H. ducreyi is available in the United States, but such testing can be performed by clinical laboratories that have developed their own PCR test and have conducted a CLIA verification study. -

Reportable Disease Surveillance in Virginia, 2013
Reportable Disease Surveillance in Virginia, 2013 Marissa J. Levine, MD, MPH State Health Commissioner Report Production Team: Division of Surveillance and Investigation, Division of Disease Prevention, Division of Environmental Epidemiology, and Division of Immunization Virginia Department of Health Post Office Box 2448 Richmond, Virginia 23218 www.vdh.virginia.gov ACKNOWLEDGEMENT In addition to the employees of the work units listed below, the Office of Epidemiology would like to acknowledge the contributions of all those engaged in disease surveillance and control activities across the state throughout the year. We appreciate the commitment to public health of all epidemiology staff in local and district health departments and the Regional and Central Offices, as well as the conscientious work of nurses, environmental health specialists, infection preventionists, physicians, laboratory staff, and administrators. These persons report or manage disease surveillance data on an ongoing basis and diligently strive to control morbidity in Virginia. This report would not be possible without the efforts of all those who collect and follow up on morbidity reports. Divisions in the Virginia Department of Health Office of Epidemiology Disease Prevention Telephone: 804-864-7964 Environmental Epidemiology Telephone: 804-864-8182 Immunization Telephone: 804-864-8055 Surveillance and Investigation Telephone: 804-864-8141 TABLE OF CONTENTS INTRODUCTION Introduction ......................................................................................................................................1 -

N-Glycan Trimming in the ER and Calnexin/Calreticulin Cycle
Neurotransmitter receptorsGABA and A postsynapticreceptor activation signal transmission Ligand-gated ion channel transport GABAGABA Areceptor receptor alpha-5 alpha-1/beta-1/gamma-2 subunit GABA A receptor alpha-2/beta-2/gamma-2GABA receptor alpha-4 subunit GABAGABA receptor A receptor beta-3 subunitalpha-6/beta-2/gamma-2 GABA-AGABA receptor; A receptor alpha-1/beta-2/gamma-2GABA receptoralpha-3/beta-2/gamma-2 alpha-3 subunit GABA-A GABAreceptor; receptor benzodiazepine alpha-6 subunit site GABA-AGABA-A receptor; receptor; GABA-A anion site channel (alpha1/beta2 interface) GABA-A receptor;GABA alpha-6/beta-3/gamma-2 receptor beta-2 subunit GABAGABA receptorGABA-A receptor alpha-2receptor; alpha-1 subunit agonist subunit GABA site Serotonin 3a (5-HT3a) receptor GABA receptorGABA-C rho-1 subunitreceptor GlycineSerotonin receptor subunit3 (5-HT3) alpha-1 receptor GABA receptor rho-2 subunit GlycineGlycine receptor receptor subunit subunit alpha-2 alpha-3 Ca2+ activated K+ channels Metabolism of ingested SeMet, Sec, MeSec into H2Se SmallIntermediateSmall conductance conductance conductance calcium-activated calcium-activated calcium-activated potassium potassium potassiumchannel channel protein channel protein 2 protein 1 4 Small conductance calcium-activatedCalcium-activated potassium potassium channel alpha/beta channel 1 protein 3 Calcium-activated potassiumHistamine channel subunit alpha-1 N-methyltransferase Neuraminidase Pyrimidine biosynthesis Nicotinamide N-methyltransferase Adenosylhomocysteinase PolymerasePolymeraseHistidine basic -
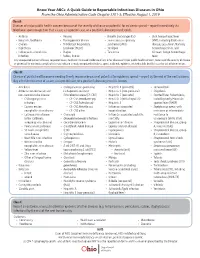
Know Your Abcs: a Quick Guide to Reportable Infectious Diseases in Ohio
Know Your ABCs: A Quick Guide to Reportable Infectious Diseases in Ohio From the Ohio Administrative Code Chapter 3701-3; Effective August 1, 2019 Class A: Diseases of major public health concern because of the severity of disease or potential for epidemic spread – report immediately via telephone upon recognition that a case, a suspected case, or a positive laboratory result exists. • Anthrax • Measles • Rubella (not congenital) • Viral hemorrhagic fever • Botulism, foodborne • Meningococcal disease • Severe acute respiratory (VHF), including Ebola virus • Cholera • Middle East Respiratory syndrome (SARS) disease, Lassa fever, Marburg • Diphtheria Syndrome (MERS) • Smallpox hemorrhagic fever, and • Influenza A – novel virus • Plague • Tularemia Crimean-Congo hemorrhagic infection • Rabies, human fever Any unexpected pattern of cases, suspected cases, deaths or increased incidence of any other disease of major public health concern, because of the severity of disease or potential for epidemic spread, which may indicate a newly recognized infectious agent, outbreak, epidemic, related public health hazard or act of bioterrorism. Class B: Disease of public health concern needing timely response because of potential for epidemic spread – report by the end of the next business day after the existence of a case, a suspected case, or a positive laboratory result is known. • Amebiasis • Carbapenemase-producing • Hepatitis B (perinatal) • Salmonellosis • Arboviral neuroinvasive and carbapenem-resistant • Hepatitis C (non-perinatal) • Shigellosis -

Communicable Diseases Weekly Report
Communicable Diseases Weekly Report Week 08, 21 February to 27 February 2021 In summary, we report: • Chancroid – one new case in a returned traveller • Rodent-borne disease risks • Novel coronavirus 2019 (COVID-19) • Summary of notifiable conditions activity in NSW For further information see NSW Health infectious diseases page. This includes links to other NSW Health infectious disease surveillance reports and a diseases data page for a range of notifiable infectious diseases. Chancroid One case of chancroid was notified this reporting week in a traveller returning from overseas. Chancroid is an acute sexually transmitted bacterial infection that causes painful genital ulcers. The condition is now rarely seen in Australia and only one other case has been notified in NSW during the past decade. Although the incidence of chancroid is decreasing globally, it is still reported in some regions within Africa, Asia, the Caribbean and South Pacific. Chancroid genital ulcer disease is a known risk factor for the transmission of HIV. The bacterium that causes chancroid, Haemophilus ducreyi, is usually transmitted through anal, oral, or vaginal sex with an infected person. After infection, one or more ulcers (sores) develop on the genitals or around the anus. Non-genital skin infections have also been reported globally, through non-sexual skin-to-skin contact with an infected person. The ulcers are usually painful, but rarely can be asymptomatic. Swelling in the groin (due to enlarged painful lymph nodes that can liquify and develop into buboes) can also occur. Other symptoms may include pain during sexual intercourse or while urinating. An infected person can spread the infection from their genital region to other parts of their body. -

Annual Summary of Communicable Disease Reported to MDH, 2003
MINNESOTA DEPARTMENT OF HEALTH DISEASE CONTROL N EWSLETTER Volume 32, Number 4 (pages 33-52) July/August 2004 Annual Summary of Communicable Diseases Reported to the Minnesota Department of Health, 2003 Introduction Minnesota Government Data Practices do not appear in Table 2 because the Assessment is a core public health Act (Section 13.38). Provisions of the influenza surveillance system is based function. Surveillance for communi- Health Insurance Portability and on reported outbreaks rather than on cable diseases is one type of ongoing Accountability Act (HIPAA) allow for individual cases. assessment activity. Epidemiologic routine communicable disease report- surveillance is the systematic collec- ing without patient authorization. Incidence rates in this report were tion, analysis, and dissemination of calculated using disease-specific health data for the planning, implemen- Since April 1995, MDH has participated numerator data collected by MDH and a tation, and evaluation of public health as one of the Emerging Infections standardized set of denominator data programs. The Minnesota Department Program (EIP) sites funded by the derived from U.S. Census data. of Health (MDH) collects disease Centers for Disease Control and Disease incidence may be categorized surveillance information on certain Prevention (CDC) and, through this as occurring within the seven-county communicable diseases for the program, has implemented active Twin Cities metropolitan area (Twin purposes of determining disease hospital- and laboratory-based surveil- Cities metropolitan area) or outside of it impact, assessing trends in disease lance for several conditions, including (Greater Minnesota). occurrence, characterizing affected selected invasive bacterial diseases populations, prioritizing disease control and food-borne diseases. Anaplasmosis efforts, and evaluating disease preven- Human anaplasmosis (HA) is the new tion strategies.