Advanced Programming in Engineering Lecture Notes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chapter 5 Dimensional Analysis and Similarity
Chapter 5 Dimensional Analysis and Similarity Motivation. In this chapter we discuss the planning, presentation, and interpretation of experimental data. We shall try to convince you that such data are best presented in dimensionless form. Experiments which might result in tables of output, or even mul- tiple volumes of tables, might be reduced to a single set of curves—or even a single curve—when suitably nondimensionalized. The technique for doing this is dimensional analysis. Chapter 3 presented gross control-volume balances of mass, momentum, and en- ergy which led to estimates of global parameters: mass flow, force, torque, total heat transfer. Chapter 4 presented infinitesimal balances which led to the basic partial dif- ferential equations of fluid flow and some particular solutions. These two chapters cov- ered analytical techniques, which are limited to fairly simple geometries and well- defined boundary conditions. Probably one-third of fluid-flow problems can be attacked in this analytical or theoretical manner. The other two-thirds of all fluid problems are too complex, both geometrically and physically, to be solved analytically. They must be tested by experiment. Their behav- ior is reported as experimental data. Such data are much more useful if they are ex- pressed in compact, economic form. Graphs are especially useful, since tabulated data cannot be absorbed, nor can the trends and rates of change be observed, by most en- gineering eyes. These are the motivations for dimensional analysis. The technique is traditional in fluid mechanics and is useful in all engineering and physical sciences, with notable uses also seen in the biological and social sciences. -
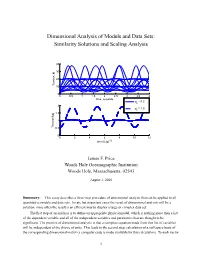
Dimensional Analysis of Models and Data Sets: Similarity Solutions and Scaling Analysis
Dimensional Analysis of Models and Data Sets: Similarity Solutions and Scaling Analysis 40 30 20 Tension, N 10 0 0 0.5 1 1.5 2 2.5 3 3.5 4 time, seconds φ = 0.2 o 2 φ = 1.0 o 1.5 1 Tension/Mg 0.5 0 0 2 4 6 8 10 12 time/(L/g)1/2 James F. Price Woods Hole Oceanographic Institution Woods Hole, Massachusetts, 02543 August 3, 2006 Summary: This essay describes a three-step procedure of dimensional analysis that can be applied to all quantitative models and data sets. In rare but important cases the result of dimensional analysis will be a solution; more often the result is an efficient way to display a large or complex data set. The first step of an analysis is to define an appropriate physical model, which is nothing more than a list of the dependent variable and all of the independent variables and parameters that are thought to be significant. The premise of dimensional analysis is that a complete equation made from this list of variables will be independent of the choice of units. This leads to the second step, calculation of a null space basis of the corresponding dimensional matrix (a computer code is made available for this calculation). To each vector 1 of the null space basis there corresponds a nondimensional variable, the number of which is less than the number of dimensional variables. The nondimensional variables are themselves a basis set and in most cases their form is not determined by dimensional analysis alone. -

Parametric Study of Magnetic Pendulum
Parametric Study of Magnetic Pendulum Author: Peter DelCioppo Persistent link: http://hdl.handle.net/2345/564 This work is posted on eScholarship@BC, Boston College University Libraries. Boston College Electronic Thesis or Dissertation, 2007 Copyright is held by the author, with all rights reserved, unless otherwise noted. Parametric Study of Magnetic Pendulum Boston College College of Arts and Sciences Honors Program Senior Thesis Project By Peter DelCioppo Advisor: Dr. Andrzej Herczynski, Physics Department Submitted May 2007 Table of Contents I. Genesis of Project…………………………………………………………………...p. 2 II Survey of Existing Literature…………………………………………………...…p. 2 III. Experimental Design……………………………………………………………...p. 3 IV. Theoretical Analysis………………………………………………………………p. 6 V. Calibration of Experimental Apparatus………………………………………….p. 8 VI. Experimental Results…………………………………………………………....p. 12 VII. Summary………………………………………………………………………..p. 15 VIII. References……………………………………………………………………...p. 17 - 1 - I. Genesis of Project This thesis project has been selected as an extension of experiments and research performed as part of the Spring 2006 Introduction to Chaos & Nonlinear Dynamics course (PH545). The current work particularly relates to experimental work analyzing the dynamics of the double well chaotic pendulum and a final project surveying the magnetic pendulum. In the experimental work, a basic understanding of the chaotic pendulum was developed, while the final project included a survey of the basic components of a magnetic pendulum system with particular -

A High-Order Boris Integrator
A high-order Boris integrator Mathias Winkela,∗, Robert Speckb,a, Daniel Ruprechta aInstitute of Computational Science, University of Lugano, Switzerland. bJ¨ulichSupercomputing Centre, Forschungszentrum J¨ulich,Germany. Abstract This work introduces the high-order Boris-SDC method for integrating the equations of motion for electrically charged particles in an electric and magnetic field. Boris-SDC relies on a combination of the Boris-integrator with spectral deferred corrections (SDC). SDC can be considered as preconditioned Picard iteration to com- pute the stages of a collocation method. In this interpretation, inverting the preconditioner corresponds to a sweep with a low-order method. In Boris-SDC, the Boris method, a second-order Lorentz force integrator based on velocity-Verlet, is used as a sweeper/preconditioner. The presented method provides a generic way to extend the classical Boris integrator, which is widely used in essentially all particle-based plasma physics simulations involving magnetic fields, to a high-order method. Stability, convergence order and conserva- tion properties of the method are demonstrated for different simulation setups. Boris-SDC reproduces the expected high order of convergence for a single particle and for the center-of-mass of a particle cloud in a Penning trap and shows good long-term energy stability. Keywords: Boris integrator, time integration, magnetic field, high-order, spectral deferred corrections (SDC), collocation method 1. Introduction Often when modeling phenomena in plasma physics, for example particle dynamics in fusion vessels or particle accelerators, an externally applied magnetic field is vital to confine the particles in the physical device [1, 2]. In many cases, such as instabilities [3] and high-intensity laser plasma interaction [4], the magnetic field even governs the microscopic evolution and drives the phenomena to be studied. -

Vibration Odes 1
Vibration ODEs 1 Vibration problems lead to differential equations with solutions that oscillate in time, typically in a damped or undamped sinusoidal fashion. Such solutions put certain demands on the numerical methods compared to other phenomena whose solutions are monotone or very smooth. Both the frequency and amplitude of the oscillations need to be accurately handled by the numerical schemes. The forthcom- ing text presents a range of different methods, from classical ones (Runge-Kutta and midpoint/Crank-Nicolson methods), to more modern and popular symplectic (ge- ometric) integration schemes (Leapfrog, Euler-Cromer, and Störmer-Verlet meth- ods), but with a clear emphasis on the latter. Vibration problems occur throughout mechanics and physics, but the methods discussed in this text are also fundamen- tal for constructing successful algorithms for partial differential equations of wave nature in multiple spatial dimensions. 1.1 Finite Difference Discretization Many of the numerical challenges faced when computing oscillatory solutions to ODEs and PDEs can be captured by the very simple ODE u00 C u D 0. This ODE is thus chosen as our starting point for method development, implementation, and analysis. 1.1.1 A Basic Model for Vibrations The simplest model of a vibrating mechanical system has the following form: u00 C !2u D 0; u.0/ D I; u0.0/ D 0; t 2 .0; T : (1.1) Here, ! and I are given constants. Section 1.12.1 derives (1.1) from physical prin- ciples and explains what the constants mean. The exact solution of (1.1)is u.t/ D I cos.!t/ : (1.2) © The Author(s) 2017 1 H.P. -

Dimensional Analysis and Modeling
cen72367_ch07.qxd 10/29/04 2:27 PM Page 269 CHAPTER DIMENSIONAL ANALYSIS 7 AND MODELING n this chapter, we first review the concepts of dimensions and units. We then review the fundamental principle of dimensional homogeneity, and OBJECTIVES Ishow how it is applied to equations in order to nondimensionalize them When you finish reading this chapter, you and to identify dimensionless groups. We discuss the concept of similarity should be able to between a model and a prototype. We also describe a powerful tool for engi- I Develop a better understanding neers and scientists called dimensional analysis, in which the combination of dimensions, units, and of dimensional variables, nondimensional variables, and dimensional con- dimensional homogeneity of equations stants into nondimensional parameters reduces the number of necessary I Understand the numerous independent parameters in a problem. We present a step-by-step method for benefits of dimensional analysis obtaining these nondimensional parameters, called the method of repeating I Know how to use the method of variables, which is based solely on the dimensions of the variables and con- repeating variables to identify stants. Finally, we apply this technique to several practical problems to illus- nondimensional parameters trate both its utility and its limitations. I Understand the concept of dynamic similarity and how to apply it to experimental modeling 269 cen72367_ch07.qxd 10/29/04 2:27 PM Page 270 270 FLUID MECHANICS Length 7–1 I DIMENSIONS AND UNITS 3.2 cm A dimension is a measure of a physical quantity (without numerical val- ues), while a unit is a way to assign a number to that dimension. -

A Position Based Approach to Ragdoll Simulation
LINKÖPING UNIVERSITY Department of Electrical Engineering Master Thesis A Position Based Approach to Ragdoll Simulation Fiammetta Pascucci LITH-ISY-EX- -07/3966- -SE June 2007 LINKÖPING UNIVERSITY Department of Electrical Engineering Master Thesis A position Based Approach to Ragdoll Simulation Fiammetta Pascucci LITH-ISY-EX- -07/3966- -SE June 2007 Supervisor: Ingemar Ragnemalm Examinator: Ingemar Ragnemalm Abstract Create the realistic motion of a character is a very complicated work. This thesis aims to create interactive animation for characters in three dimen- sions using position based approach. Our character is pictured from ragdoll, which is a structure of system particles where all particles are linked by equidistance con- straints. The goal of this thesis is observed the fall in the space of our ragdoll after creating all constraints, as structure, contact and environment constraints. The structure constraint represents all joint constraints which have one, two or three Degree of Freedom (DOF). The contact constraints are represented by collisions between our ragdoll and other objects in the space. Finally, the environment constraints are represented by means of the wall con- straint. The achieved results allow to have a realist fall of our ragdoll in the space. Keywords: Particle System, Verlet’s Integration, Skeleton Animation, Joint Con- straint, Environment Constraint, Skinning, Collision Detection. Acknowledgments I would to thanks all peoples who aid on making my master thesis work. In particular, thanks a lot to the person that most of all has believed in me. This person has always encouraged to go forward, has always given a lot love and help me in difficult movement. -

Exponential Integration for Hamiltonian Monte Carlo
Exponential Integration for Hamiltonian Monte Carlo Wei-Lun Chao1 [email protected] Justin Solomon2 [email protected] Dominik L. Michels2 [email protected] Fei Sha1 [email protected] 1Department of Computer Science, University of Southern California, Los Angeles, CA 90089 2Department of Computer Science, Stanford University, 353 Serra Mall, Stanford, California 94305 USA Abstract butions that generate random walks in sample space. A fraction of these walks aligned with the target distribution is We investigate numerical integration of ordinary kept, while the remainder is discarded. Sampling efficiency differential equations (ODEs) for Hamiltonian and quality depends critically on the proposal distribution. Monte Carlo (HMC). High-quality integration is For this reason, designing good proposals has long been a crucial for designing efficient and effective pro- subject of intensive research. posals for HMC. While the standard method is leapfrog (Stormer-Verlet)¨ integration, we propose For distributions over continuous variables, Hamiltonian the use of an exponential integrator, which is Monte Carlo (HMC) is a state-of-the-art sampler using clas- robust to stiff ODEs with highly-oscillatory com- sical mechanics to generate proposal distributions (Neal, ponents. This oscillation is difficult to reproduce 2011). The method starts by introducing auxiliary sam- using leapfrog integration, even with carefully pling variables. It then treats the negative log density of selected integration parameters and precondition- the variables’ joint distribution as the Hamiltonian of parti- ing. Concretely, we use a Gaussian distribution cles moving in sample space. Final positions on the motion approximation to segregate stiff components of trajectories are used as proposals. the ODE. -

Simple Quantitative Tests to Validate Sampling from Thermodynamic
Simple quantitative tests to validate sampling from thermodynamic ensembles Michael R. Shirts∗ Department of Chemical Engineering, University of Virginia, VA 22904 E-mail: [email protected] Abstract It is often difficult to quantitatively determine if a new molecular simulation algorithm or software properly implements sampling of the desired thermodynamic ensemble. We present some simple statistical analysis procedures to allow sensitive determination of whether a de- sired thermodynamic ensemble is properly sampled. We demonstrate the utility of these tests for model systems and for molecular dynamics simulations in a range of situations, includ- ing constant volume and constant pressure simulations, and describe an implementation of the tests designed for end users. 1 Introduction Molecular simulations, including both molecular dynamics (MD) and Monte Carlo (MC) tech- arXiv:1208.0910v1 [cond-mat.stat-mech] 4 Aug 2012 niques, are powerful tools to study the properties of complex molecular systems. When used to specifically study thermodynamics of such systems, rather than dynamics, the primary goal of molecular simulation is to generate uncorrelated samples from the appropriate ensemble as effi- ciently as possible. This simulation data can then be used to compute thermodynamic properties ∗To whom correspondence should be addressed 1 of interest. Simulations of several different ensembles may be required to simulate some thermo- dynamic properties, such as free energy differences between states. An ever-expanding number of techniques have been proposed to perform more and more sophisticated sampling from com- plex molecular systems using both MD and MC, and new software tools are continually being introduced in order to implement these algorithms and to take advantage of advances in hardware architecture and programming languages. -

Dimensional Analysis and Modeling
cen72367_ch07.qxd 10/29/04 2:27 PM Page 269 CHAPTER DIMENSIONAL ANALYSIS 7 AND MODELING n this chapter, we first review the concepts of dimensions and units. We then review the fundamental principle of dimensional homogeneity, and OBJECTIVES Ishow how it is applied to equations in order to nondimensionalize them When you finish reading this chapter, you and to identify dimensionless groups. We discuss the concept of similarity should be able to between a model and a prototype. We also describe a powerful tool for engi- ■ Develop a better understanding neers and scientists called dimensional analysis, in which the combination of dimensions, units, and of dimensional variables, nondimensional variables, and dimensional con- dimensional homogeneity of equations stants into nondimensional parameters reduces the number of necessary ■ Understand the numerous independent parameters in a problem. We present a step-by-step method for benefits of dimensional analysis obtaining these nondimensional parameters, called the method of repeating ■ Know how to use the method of variables, which is based solely on the dimensions of the variables and con- repeating variables to identify stants. Finally, we apply this technique to several practical problems to illus- nondimensional parameters trate both its utility and its limitations. ■ Understand the concept of dynamic similarity and how to apply it to experimental modeling 269 cen72367_ch07.qxd 10/29/04 2:27 PM Page 270 270 FLUID MECHANICS Length 7–1 ■ DIMENSIONS AND UNITS 3.2 cm A dimension is a measure of a physical quantity (without numerical val- ues), while a unit is a way to assign a number to that dimension. -

Transport Properties of Fluids: Symplectic Integrators and Their
Fluid Phase Equilibria 183–184 (2001) 351–361 Transport properties of fluids: symplectic integrators and their usefulness J. Ratanapisit a,b, D.J. Isbister c, J.F. Ely b,∗ a Chemical Engineering and Petroleum Refining Department, Colorado School of Mines, Golden, CO 80401, USA b Department of Chemical Engineering, Prince of Songla University, Hat-Yai, Songkla 90110, Thailand c School of Physics, University College, University of New South Wales, ADFA, Canberra, ACT 2600, Australia Abstract An investigation has been carried out into the effectiveness of using symplectic/operator splitting generated algorithms for the evaluation of transport coefficients in Lennard–Jones fluids. Equilibrium molecular dynamics is used to revisit the Green–Kubo calculation of these transport coefficients through integration of the appropriate correlation functions. In particular, an extensive series of equilibrium molecular dynamic simulations have been per- formed to investigate the accuracy, stability and efficiency of second-order explicit symplectic integrators: position Verlet, velocity Verlet, and the McLauchlan–Atela algorithms. Comparisons are made to nonsymplectic integrators that include the fourth-order Runge–Kutta and fourth-order Gear predictor–corrector methods. These comparisons were performed based on several transport properties of Lennard–Jones fluids: self-diffusion, shear viscosity and thermal conductivity. Because transport properties involve long time simulations to obtain accurate evaluations of their numerical values, they provide an excellent basis to study the accuracy and stability of the SI methods. To our knowledge, previous studies on the SIs have only looked at the thermodynamic energy using a simple model fluid. This study presents realistic, but perhaps the simplest simulations possible to test the effect of the integrators on the three main transport properties. -

Verlet with Collisions for Mass Spring Model Simulations
Verlet with Collisions for Mass Spring Model Simulations Maciej Kot1 and Hiroshi Nagahashi2 1Imaging Science and Engineering Laboratory, Tokyo Institute of Technology, Tokyo, Japan 2Laboratory for Future Interdisciplinary Research of Science and Technology, Tokyo Institute of Technology, Tokyo, Japan Keywords: Mass Spring Models, Deformable Objects, Collisions, Verlet, Friction. Abstract: In this paper we study the problem of the interaction of soft bodies modeled with mass spring models (MSM) and static elements of the environment. We show that in such setup it is possible to couple standard time evolution of MSMs with collision responses in a way, that does not require complex processing for multi collision situations while successfully preventing object inter-penetration. Moreover we show how to achieve similar energy dissipation for models with different resolutions when the friction is present. 1 INTRODUCTION ulation and we show how to couple it with collision responses. One of our goals is to design a collision Mass spring models are a popular choice for the rep- handling technique which preserves the energy of the resentation of soft bodies in computer graphics and system (affects stability of the simulation), prevents virtual reality applications. For a long time they were object penetration, and if the friction is present, obeys preferred over the finite element method (FEM) in the Newton’s law of dry friction. We achieve this goal real time applications, as they tend to offer better completely for collisions between MSM and static el- computational efficiency (although recent FEM im- ements of the environment and partially for MSM- plementations are quite fast as well (Sin et al., 2013)), MSM collisions; in case of MSM-MSM collisions our but they are believed to be less accurate in terms of method does not preserve energy in certain situations, physical plausibility (Nealen et al., 2006).