Animal Reproduction Science 208 (2019) 106078
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of Synonymous Codon Usage Patterns in Sixty-Four Different Bivalve Species
Analysis of synonymous codon usage patterns in sixty-four diVerent bivalve species Marco Gerdol1, Gianluca De Moro1, Paola Venier2 and Alberto Pallavicini1 1 Department of Life Sciences, University of Trieste, Trieste, Italy 2 Department of Biology, University of Padova, Padova, Italy ABSTRACT Synonymous codon usage bias (CUB) is a defined as the non-random usage of codons encoding the same amino acid across diVerent genomes. This phenomenon is common to all organisms and the real weight of the many factors involved in its shaping still remains to be fully determined. So far, relatively little attention has been put in the analysis of CUB in bivalve mollusks due to the limited genomic data available. Taking advantage of the massive sequence data generated from next generation sequencing projects, we explored codon preferences in 64 diVerent species pertaining to the six major evolutionary lineages in Bivalvia. We detected remarkable diVerences across species, which are only partially dependent on phylogeny. While the intensity of CUB is mild in most organisms, a heterogeneous group of species (including Arcida and Mytilida, among the others) display higher bias and a strong preference for AT-ending codons. We show that the relative strength and direction of mutational bias, selection for translational eYciency and for translational accuracy contribute to the establishment of synonymous codon usage in bivalves. Although many aspects underlying bivalve CUB still remain obscure, we provide for the first time an overview of this phenomenon -

Biogeographical Homogeneity in the Eastern Mediterranean Sea. II
Vol. 19: 75–84, 2013 AQUATIC BIOLOGY Published online September 4 doi: 10.3354/ab00521 Aquat Biol Biogeographical homogeneity in the eastern Mediterranean Sea. II. Temporal variation in Lebanese bivalve biota Fabio Crocetta1,*, Ghazi Bitar2, Helmut Zibrowius3, Marco Oliverio4 1Stazione Zoologica Anton Dohrn, Villa Comunale, 80121, Napoli, Italy 2Department of Natural Sciences, Faculty of Sciences, Lebanese University, Hadath, Lebanon 3Le Corbusier 644, 280 Boulevard Michelet, 13008 Marseille, France 4Dipartimento di Biologia e Biotecnologie ‘Charles Darwin’, University of Rome ‘La Sapienza’, Viale dell’Università 32, 00185 Roma, Italy ABSTRACT: Lebanon (eastern Mediterranean Sea) is an area of particular biogeographic signifi- cance for studying the structure of eastern Mediterranean marine biodiversity and its recent changes. Based on literature records and original samples, we review here the knowledge of the Lebanese marine bivalve biota, tracing its changes during the last 170 yr. The updated checklist of bivalves of Lebanon yielded a total of 114 species (96 native and 18 alien taxa), accounting for ca. 26.5% of the known Mediterranean Bivalvia and thus representing a particularly poor fauna. Analysis of the 21 taxa historically described on Lebanese material only yielded 2 available names. Records of 24 species are new for the Lebanese fauna, and Lioberus ligneus is also a new record for the Mediterranean Sea. Comparisons between molluscan records by past (before 1950) and modern (after 1950) authors revealed temporal variations and qualitative modifications of the Lebanese bivalve fauna, mostly affected by the introduction of Erythraean species. The rate of recording of new alien species (evaluated in decades) revealed later first local arrivals (after 1900) than those observed for other eastern Mediterranean shores, while the peak in records in conjunc- tion with our samplings (1991 to 2010) emphasizes the need for increased field work to monitor their arrival and establishment. -
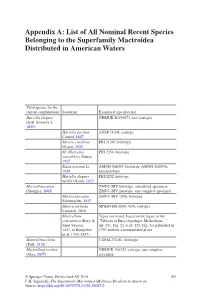
List of All Nominal Recent Species Belonging to the Superfamily Mactroidea Distributed in American Waters
Appendix A: List of All Nominal Recent Species Belonging to the Superfamily Mactroidea Distributed in American Waters Valid species (in the current combination) Synonym Examined type material Harvella elegans NHMUK 20190673, two syntypes (G.B. Sowerby I, 1825) Harvella pacifica ANSP 51308, syntype Conrad, 1867 Mactra estrellana PRI 21265, holotype Olsson, 1922 M. (Harvella) PRI 2354, holotype sanctiblasii Maury, 1925 Raeta maxima Li, AMNH 268093, lectotype; AMNH 268093a, 1930 paralectotype Harvella elegans PRI 2252, holotype tucilla Olsson, 1932 Mactrellona alata ZMUC-BIV, holotype, articulated specimen; (Spengler, 1802) ZMUC-BIV, paratype, one complete specimen Mactra laevigata ZMUC-BIV 1036, holotype Schumacher, 1817 Mactra carinata MNHN-IM-2000-7038, syntypes Lamarck, 1818 Mactrellona Types not found, based on the figure of the concentrica (Bory de “Tableau of Encyclopedique Methodique…” Saint Vincent, (pl. 251, Fig. 2a, b, pl. 252, Fig. 2c) published in 1827, in Bruguière 1797 without a nomenclatorial act et al. 1791–1827) Mactrellona clisia USNM 271481, holotype (Dall, 1915) Mactrellona exoleta NHMUK 196327, syntype, one complete (Gray, 1837) specimen © Springer Nature Switzerland AG 2019 103 J. H. Signorelli, The Superfamily Mactroidea (Mollusca:Bivalvia) in American Waters, https://doi.org/10.1007/978-3-030-29097-9 104 Appendix A: List of All Nominal Recent Species Belonging to the Superfamily… Valid species (in the current combination) Synonym Examined type material Lutraria ventricosa MCZ 169451, holotype; MCZ 169452, paratype; -

Fossil Bivalves and the Sclerochronological Reawakening
Paleobiology, 2021, pp. 1–23 DOI: 10.1017/pab.2021.16 Review Fossil bivalves and the sclerochronological reawakening David K. Moss* , Linda C. Ivany, and Douglas S. Jones Abstract.—The field of sclerochronology has long been known to paleobiologists. Yet, despite the central role of growth rate, age, and body size in questions related to macroevolution and evolutionary ecology, these types of studies and the data they produce have received only episodic attention from paleobiologists since the field’s inception in the 1960s. It is time to reconsider their potential. Not only can sclerochrono- logical data help to address long-standing questions in paleobiology, but they can also bring to light new questions that would otherwise have been impossible to address. For example, growth rate and life-span data, the very data afforded by chronological growth increments, are essential to answer questions related not only to heterochrony and hence evolutionary mechanisms, but also to body size and organism ener- getics across the Phanerozoic. While numerous fossil organisms have accretionary skeletons, bivalves offer perhaps one of the most tangible and intriguing pathways forward, because they exhibit clear, typically annual, growth increments and they include some of the longest-lived, non-colonial animals on the planet. In addition to their longevity, modern bivalves also show a latitudinal gradient of increasing life span and decreasing growth rate with latitude that might be related to the latitudinal diversity gradient. Is this a recently developed phenomenon or has it characterized much of the group’s history? When and how did extreme longevity evolve in the Bivalvia? What insights can the growth increments of fossil bivalves provide about hypotheses for energetics through time? In spite of the relative ease with which the tools of sclerochronology can be applied to these questions, paleobiologists have been slow to adopt sclerochrono- logical approaches. -

Revision of the Magellanic Mactridae Lamarck, 1809 (Bivalvia: Heterodonta)
Zootaxa 2757: 47–67 (2011) ISSN 1175-5326 (print edition) www.mapress.com/zootaxa/ Article ZOOTAXA Copyright © 2011 · Magnolia Press ISSN 1175-5334 (online edition) Revision of the Magellanic Mactridae Lamarck, 1809 (Bivalvia: Heterodonta) JAVIER H. SIGNORELLI1 & GUIDO PASTORINO2 1Biología y Manejo de Recursos Acuáticos, LARBIM - CENPAT – CONICET. Bvd. Brown 2915, U9120ACD, Puerto Madryn, Chubut, Argentina. E-mail: [email protected] 2Museo Argentino de Ciencias Naturales “Bernardino Rivadavia”, Av. Ángel Gallardo 470 C1405DJR, Buenos Aires, Argentina. Abstract The worldwide family Mactridae Lamarck, 1809, is well represented in the southwestern Atlantic Ocean. Literature re- search revealed 22 living nominal species linked to Magellanic forms. Within this context, a taxonomic revision of the species belonging to the family Mactridae that inhabit the Magellanic area was carried out. The analysis was done by using morphological characters of the shell and describing the mantle cavity organs. After extensive fieldwork from Peninsula Valdés to Tierra del Fuego province, seven valid living species were confirmed. Synonymies of those valid forms have been updated, as have their geographic distributions. Redescriptions of the species here considered as valid, i.e.: Mactra fuegiensis Smith, 1905, Mulinia bicolor Gray, 1837, Mulinia byronensis Gray, 1837, Mulinia edulis (King & Broderip, 1832), Mulinia exalbida Gray, 1837, Mulinia levicardo (Smith 1881) and Darina solenoides (King & Broderip, 1832) are provided. Key words: Mactra, Mulinia, Darina, Taxonomy, Southwestern Atlantic Introduction The family Mactridae, known in North America since the lower Cretaceous (Skelton & Benton, 1993), includes five subfamilies: Mactrinae Lamarck, 1809, Lutrariinae H. Adams & A. Adams (1856), Kymatoxinae Stenzel & Krause, 1957, Zenatinae Dall, 1895, and Tanysiphoninae Scarlato & Starobogatov, in Nevesskaja et al., 1971. -

Bycatch in Bivalve Fisheries of Algarve
Mariana Anjos BYCATCH IN BIVALVE FISHERIES OF ALGARVE UNIVERSIDADE DO ALGARVE Faculdade de Ciências e Tecnologia 2016 Mariana Anjos BYCATCH IN BIVALVE FISHERIES OF ALGARVE Mestrado em Biologia Marinha Trabalho efetuado sob a orientação de: Miguel Gaspar, Ph.D. Karim Erzini, Ph.D. UNIVERSIDADE DO ALGARVE Faculdade de Ciências e Tecnologia 2016 Bycatch in Bivalve fisheries of Algarve DECLARAÇÃO DE AUTORIA DE TRABALHO Declaro ser a autora deste trabalho, que é original e inédito. Autores e trabalhos consultados estão devidamente citados no texto e constam da listagem de referências incluída. Copyright © Mariana Anjos 2016 A Universidade do Algarve reserva para si o direito, em conformidade com o disposto no Código do Direito de Autor e dos Direitos Conexos, de arquivar, reproduzir e publicar a obra, independentemente do meio utilizado, bem como de a divulgar através de repositórios científicos e de admitir a sua cópia e distribuição para fins meramente educacionais ou de investigação e não comerciais, conquanto seja dado o devido crédito ao autor e editor respetivos. III AGRADECIMENTOS Obrigada ao Dr. Miguel Gaspar, Dr. Karim Erzini, Fábio Pereira e Dr. Paulo Vasconcelos pela ajuda imprescindível que me deram ao longo desta tese de mestrado. A todos os que no IPMA ajudaram a triar quantidades imensas de amêijoa, um grande obrigada. Aos pescadores da Renovadora e Cláudia Marina sem os quais esta tese não teria sido possível. Obrigada mãe, pai, tio, André e Ana pela paciência e apoio durante a minha ausência nestes meses de trabalho. À minha irmã Joana por estar sempre presente e por ter paciência quando a paciência a todos os outros faltava. -

Chemical Taxonomy of the Hinge-Ligament Proteins of Bivalves According to Their Amino Acid Compositions
Biochem. J. (1987) 242, 505-510 (Printed in Great Britain) 505 Chemical taxonomy of the hinge-ligament proteins of bivalves according to their amino acid compositions Yasuo KIKUCHI* and Nobuo TAMIYA Department of Chemistry, Faculty of Science, Tohoku University Aobayama, Sendai 980, Japan The proteins in the hinge ligaments of molluscan bivalves were subjected to chemotaxonomic studies according to their amino acid compositions. The hinge-ligament protein is a new class of structure proteins, and this is the first attempt to introduce chemical taxonomy into the systematics of bivalves. The hinge-ligament proteins from morphologically close species, namely mactra (superfamily Mactracea) or scallop (family Pectinidae) species, showed high intraspecific homology in their compositions. On the other hand, inconsistent results were obtained with two types of ligament proteins in pearl oyster species (genus Pinctada). The results of our chemotaxonomic analyses were sometimes in good agreement with the morphological classifications and sometimes inconsistent, implying a complicated phylogenetic relationship among the species. INTRODUCTION (1982). The ligaments were removed from the shells and The two shells of molluscan bivalves are connected dried in vacuo. with each other by the organic ligament at the hinge. The Amino acid compositions of the ligament proteins hinge ligament is elastic and functions to open the shells: Small pieces (about 2 mg) of the ligament were heated the ligament is strained when the shells are closed by the in 6 M-HCI (0.3 ml) at 105 °C for 24 h in vacuo. The adductor muscles, and when the muscles relax the amino acids in the hydrolysate were analysed with a JLC spring-like action ofthe elastic ligament opens the valves. -

External Ca2+ Is Predominantly Used for Cytoplasmic and Nuclear Ca2+ Increases in Fertilized Oocytes of the Marine Bivalve Mactra Chinensis
Research Article 367 External Ca2+ is predominantly used for cytoplasmic and nuclear Ca2+ increases in fertilized oocytes of the marine bivalve Mactra chinensis Ryusaku Deguchi1,* and Masaaki Morisawa2 1Department of Biology, Miyagi University of Education, Aoba-ku, Sendai, Miyagi 980-0845, Japan 2Misaki Marine Biological Station, the University of Tokyo, Miura, Kanagawa 238-0225, Japan *Author for correspondence (e-mail: [email protected]) Accepted 15 October 2002 Journal of Cell Science 116, 367-376 © 2003 The Company of Biologists Ltd doi:10.1242/jcs.00221 Summary Oocytes of the marine bivalve Mactra chinensis are source. In contrast to the situation observed at fertilization, spawned and arrested at the germinal vesicle stage (first an oocyte artificially stimulated with serotonin (5- meiotic prophase) until fertilization, without undergoing a hydroxytryptamine, 5-HT) displayed repetitive Ca2+ process called oocyte maturation. As is the case of other transients, each of which started from one cortical region animals, a fertilized oocyte of the bivalve displays increases and propagated across the oocyte as a Ca2+ wave. The 5- in intracellular free Ca2+. We have clarified here the HT-induced Ca2+ transients persisted even in the absence 2+ spatiotemporal patterns and sources of the intracellular of external Ca . Experiments with caged Ins(1,4,5)P3 2+ 2+ Ca changes at fertilization. Shortly after insemination, revealed that Ca release from Ins(1,4,5)P3-sensitive stores increased Ca2+ simultaneously appeared at the whole is another pathway that is sufficient to trigger meiosis cortical region of the oocyte and spread inwardly to the reinitiation from the first prophase. -

IMPACTS of SELECTIVE and NON-SELECTIVE FISHING GEARS on the INLAND WATERS of BANGLADESH
A checklist of molluscans inhabiting Bandri Beach along the Jiwani coast, Balochistan, Pakistan Item Type article Authors Ghani, Abdul; Afsar, Nuzhat; Moazzam, Muhammad Download date 26/09/2021 00:44:50 Link to Item http://hdl.handle.net/1834/40825 Pakistan Journal of Marine Sciences, Vol. 27(1), 61-71, 2018. A CHECKLIST OF MOLLUSCANS INHABITING BANDRI BEACH ALONG THE JIWANI COAST, BALOCHISTAN, PAKISTAN Abdul Ghani, Nuzhat Afsar and Muhammad Moazzam Institute of Marine Science, University of Karachi, Karachi-75270, Pakistan. (AG, NA); WWF, Karachi office, Bungalow # 46/K, Block 6, P.E.C.H.S, Shahrah-e-Faisal, Karachi (MM). email: [email protected]; [email protected] ABSTRACT: Main object of the study was to record the composition and diversity of intertidal molluscan species of the Bandri Beach along the Jiwani coast, Balochistan to develop baseline data information which could be helpful in future conservation perspective. The study revealed the presence of ninety eight (98) species comprising of sixty eight (68) gastropods, twenty six (26) bivalves, two (2) scaphopods, one (1) Polyplacophora and one (1) Cephalopod species at two selected points of the Bandri Beach, Jiwani coast. Among these molluscan species, members of cerithids, trochid Umbonium vestairium, bivalve Branchidontes variabilis and oyster Crassostrea madrasensis were found in abundance. Study presents the first report on the occurrence of molluscan species in the area. KEYWORDS: Molluscs, checklist, Bandri Beach, Jiwani, Balochistan coast, Pakistan. INTRODUCTION Molluscan species of Pakistan coast especially those found along the Sindh coast have been studied extensively and several authors have published papers on species diversity, distribution, and abundance (Burney and Barkati, 1995; Nasreen et al., 2000; Rahman and Barkati, 2004; Afsar et al., 2012; Rahman and Barkati, 2012; Afsar et al., 2013 a,b), as compared to molluscan abunadance and distribution studies along Balochistan coast. -

Mulinia Lateralis (Say, 1822) in Europe J
Craeymeersch et al. Marine Biodiversity Records (2019) 12:5 https://doi.org/10.1186/s41200-019-0164-7 MARINE RECORD Open Access First records of the dwarf surf clam Mulinia lateralis (Say, 1822) in Europe J. A. Craeymeersch1* , M. A. Faasse2,3, H. Gheerardyn2, K. Troost1, R. Nijland5 , A. Engelberts4 , K. J. Perdon1, D. van den Ende1 and J. van Zwol1 Abstract This paper reports the first records of the dwarf surf clam Mulinia lateralis (Say, 1822) outside its native area, which is the western Atlantic Ocean, ranging from theGulfofStLawrencetotheGulfofMexico.In2017 and 2018 specimens were found in the Dutch coastal waters (North Sea), in the Wadden Sea and in the Westerschelde estuary, in densities of up to almost 6000 individuals per square meter. In view of its ecology and distributional range in the native area M. lateralis has the potential to become an invasive species. Its ability to quickly colonize defaunated areas, its high fecundity and short generation time, its tolerance for anoxia and temperature extremes and its efficient exploitation of the high concentrations of phytoplankton and natural seston at the sediment-water interface may bring it into competition with native species for food and space. Keywords: Mulinia lateralis, Bivalvia, Marine, North Sea, Invasive, Competition Introduction shipping (Port of Rotterdam) and aquaculture (Oos- For at least half a century, the process and the envir- terschelde estuary) activities in the North Sea region onmental, economic and social impacts of invasions (see also Wolff 2005). The most successful taxa re- of non-indigenous species are a focus of ecological garding introduction and immigration are poly- research (Elton 1958; Rilov and Crooks 2009; chaetes, bivalves and amphipods (Reise et al. -

Cenozpic Pectinids of Alaska, Iceland, and Other Northern Regions
Cenozpic Pectinids of Alaska, Iceland, and Other Northern Regions GEOLOGICAL SURVEY PROFESSIONAL PAPER 553 Cenozoic Pectinids of Alaska, Iceland, and Other Northern Regions By F. STEARNS MAcNEIL GEOLOGICAL SURVEY PROFESSIONAL PAPER 553 Evolution of indigenous Alaskan pectinid stocks^ of migrant stocks from other areas in the Pacific region^ and of boreal Pacific species that subsequently migrated to the Arctic and northern Atlantic regions UNITED STATES GOVERNMENT PRINTING OFFICE, WASHINGTON : 1967 UNITED STATES DEPARTMENT OF THE INTERIOR STEW ART L. UDALL, Secretary GEOLOGICAL SURVEY William T. Pecora, Director Library of Congress catalog-card No. GS 66-321 For sale by the Superintendent of Documents, U.S. Government Printing Office Washington, D.C. 20402 - Price $1.00 (paper cover) CONTENTS Page Systematic paleontology Continued Abstract_ ________________________________________ 1 Genus CMamys Roeding Continued Introduction. ______________________________________ 1 Subgenus CMamys Roeding Continued Page Pectinidae as guide fossils__________________________ 1 Group of C. (C.) rubida (Hinds)..________ 21 Pectinidae of Alaska._______________________________ 2 Chlamys (Chlamys) rubida (Hinds)____ 21 Migration routes__________________________________ 3 Chlamys (Chlamys) rubida jordani (Ar Systematic concept.________________________________ 4 nold) 22 Systematic paleontology.____________________________ 5 Chlamys (Chlamys) rubida hindsii (Car Genus Delectopecten Stewart______________________ 5 penter) __________________________ 23 Delectopecten -

Shelled Molluscs - Berthou P., Poutiers J.-M., Goulletquer P., Dao J.C
FISHERIES AND AQUACULTURE – Vol. II - Shelled Molluscs - Berthou P., Poutiers J.-M., Goulletquer P., Dao J.C. SHELLED MOLLUSCS Berthou P. Institut Français de Recherche pour l'Exploitation de la Mer, Plouzané, France Poutiers J.-M. Muséum National d’Histoire Naturelle, Paris, France Goulletquer P. Institut Français de Recherche pour l'Exploitation de la Mer, La Tremblade, France Dao J.C. Institut Français de Recherche pour l'Exploitation de la Mer, Plouzané, France Keywords: bivalves, gastropods, fisheries, aquaculture, biology, fishing gears, management Contents 1. Introduction 1.1. Uses of Shellfish: An Overview 1.2. Production 2. Species and Fisheries 2.1. Diversity of Species 2.1.1. Edible Species 2.1.2. Shellfish Species Not Used as Food 2.2. Shelled Molluscs Fisheries 2.2.1. Gastropods 2.2.2. Oysters 2.2.3. Mussels 2.2.4. Scallops 2.2.5. Clams 2.3. Shelled Molluscs Cultivation 2.3.1. Gastropods 2.3.2. Oysters 2.3.3. Mussels 2.3.4. ScallopsUNESCO – EOLSS 2.3.5. Clams 3. Harvesting andSAMPLE Cultivation Techniques CHAPTERS 3.1. Harvesting 3.2. Cultivation techniques 4. Biology 4.1. General Ecology 4.2. Growth 4.3. Reproduction 4.4. Larval Stage in Relation to Dispersal and Stock Abundance 4.5. Migration 5. Stock Assessment and Management Approaches ©Encyclopedia of Life Support Systems (EOLSS) FISHERIES AND AQUACULTURE – Vol. II - Shelled Molluscs - Berthou P., Poutiers J.-M., Goulletquer P., Dao J.C. 5.1. Stock Assessment 5.2. Management Strategies 6. Issues for the Future Bibliography Biographical Sketches Summary Shelled molluscs are comprised of bivalves and gastropods.