Mechanisms and Consequences of Epigenetic Inheritance Following Parental Preconception Alcohol Exposure
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Introduction to Genetics Bios 225
INTRODUCTION TO GENETICS BIOS 225 Course Description This course introduces the student to the basic concepts of inheritance, populations, mutations, and techniques used to assess each of these. Credit: 2 credits Repeatable: No Course Structure The course will be presented in different formats: Lectures with PowerPoints, self-directed learning, discussions and student assignments etc. Competencies This course emphasizes competencies to enhance skills essential for a future health care professional. • Knowledge o Demonstrate content knowledge and skills in foundational courses required by biomedical professionals o Demonstrate information literacy o Demonstrate quantitative reasoning o Demonstrate longitudinal learning through coursework • Critical Thinking o Develop the skills of self-reflection and peer assessment to improve personal performance. o Demonstrate the ability to analyze literature and written material o Demonstrate the ability to distinguish between well-reasoned and poorly reasoned arguments • Communication Skills o Demonstrate effective presentation skills to faculty and peers. o Demonstrate effective listening skills o Demonstrate effective written communication 1 Objectives: Upon completion of BIOS 225 course, the student should be able to describe: • The structure and function of purines, pyrimidines, nucleosides and nucleotides • The structure and functions of nucleic acids (DNA and RNA) • The chromosome anatomy and human karyotypes • The concepts of prokaryotic and eukaryotic DNA replication • The concepts of prokaryotic and eukaryotic RNA transcription and post-transcriptional modifications • The concepts of prokaryotic and eukaryotic protein translation and post-translational modifications • The regulations of prokaryotic and eukaryotic gene expressions • The process of genomic, chromosomal and gene mutations; and its repair mechanism • The Mendel’ hypothesis and molecular mechanisms of genetic inheritance Schedule: Dates and times to be posted at the beginning of the term on the online calendar. -

Animal Traditions: Behavioural Inheritance in Evolution
Animal Traditions: Behavioural Inheritance in Evolution Eytan Avital and Eva Jablonka CAMBRIDGE UNIVERSITY PRESS Animal Traditions Behavioural Inheritance in Evolution Animal Traditions maintains that the assumption that the selection of genes supplies both a sufficient explanation of the evolution of behav- iour and a true description of its course is, despite its almost univer- sal acclaim, wrong. Eytan Avital and Eva Jablonka contend that evolutionary explanations must take into account the well-established fact that, in mammals and birds, the transfer of learnt information across generations is both ubiquitous and indispensable. The introduc- tion of the behavioural inheritance system into the Darwinian explanatory scheme enables the authors to offer new interpretations for common behaviours such as maternal behaviours, behavioural conflicts within families, adoption and helping. This approach offers a richer view of heredity and evolution, integrates developmental and evolutionary processes, suggests new lines for research and provides a constructive alternative to both the selfish gene and meme views of the world. It will make stimulating reading for all those interested in evolutionary biology, sociobiology, behavioural ecology and psychology. eytan avital is a lecturer in Zoology in the Department of Natural Sciences at David Yellin College of Education in Jerusalem. He is a highly experienced field biologist, and has written one zoology text and edited several others on zoology and evolution for the Israel Open university. eva jablonka is a senior lecturer in the Cohn Institute for the History and Philosophy of Science and Ideas, at Tel-Aiv University. She is the author of three books on heredity and evolution, most recently Epigenetic Inheritance and Evolution with Marion Lamb. -

WO 2012/174282 A2 20 December 2012 (20.12.2012) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2012/174282 A2 20 December 2012 (20.12.2012) P O P C T (51) International Patent Classification: David [US/US]; 13539 N . 95th Way, Scottsdale, AZ C12Q 1/68 (2006.01) 85260 (US). (21) International Application Number: (74) Agent: AKHAVAN, Ramin; Caris Science, Inc., 6655 N . PCT/US20 12/0425 19 Macarthur Blvd., Irving, TX 75039 (US). (22) International Filing Date: (81) Designated States (unless otherwise indicated, for every 14 June 2012 (14.06.2012) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, English (25) Filing Language: CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, Publication Language: English DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, KR, (30) Priority Data: KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, 61/497,895 16 June 201 1 (16.06.201 1) US MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, 61/499,138 20 June 201 1 (20.06.201 1) US OM, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SC, SD, 61/501,680 27 June 201 1 (27.06.201 1) u s SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, 61/506,019 8 July 201 1(08.07.201 1) u s TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. -
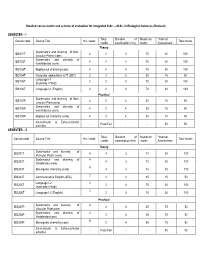
Detailed Course Matrix and Scheme of Evaluation for Integrated B.Sc. – M.Sc. in Biological Sciences (Revised) SEMESTER –
Detailed course matrix and scheme of evaluation for Integrated B.Sc. – M.Sc. in Biological Sciences (Revised) SEMESTER – I Total Duration of Maximum *Internal Course code Course Title Hrs / week Total marks credits examination (hrs) marks Assessment Theory Systematics and diversity of Non- IBS101T 4 4 3 70 30 100 vascular Plants (core) Systematics and diversity of IBS102T 4 4 3 70 30 100 Invertebrates (core) IBS103T Biophysical chemistry(core) 4 4 3 70 30 100 IBS104T Computer applications & IT (SEC) 2 2 2 35 15 50 Language I-1 IBS105T 2 2 3 70 30 100 (Kannada / Hindi) IBS106T Language I-1 (English) 2 2 3 70 30 100 Practical Systematics and diversity of Non- IBS107P 4 2 4 35 15 50 vascular Plant (core) Systematics and diversity of IBS108P 4 2 4 35 15 50 Invertebrates (core) IBS109P Biophysical chemistry (core) 4 2 4 35 15 50 Co-curricular & Extra-curricular - Pass/Fail - - 50 50 activities SEMESTER – II Total Duration of Maximum *Internal Course code Course Title Hrs / week Total marks credits examination (hrs) marks Assessment Theory Systematics and diversity of IBS201T 4 4 3 70 30 100 Vascular Plants (core) Systematics and diversity of 4 IBS202T 4 3 70 30 100 Vertebrates (core) 4 IBS203T Bio-organic chemistry (core) 4 3 70 30 100 2 IBS204T Communicative English (AEC) 2 2 35 15 50 Language I-2 2 IBS205T 2 3 70 30 100 (Kannada / Hindi) 2 IBS206T Language I-2 (English) 2 3 70 30 100 Practical Systematics and diversity of 4 IBS207P 2 4 35 15 50 Vascular Plant(core) Systematics and diversity of 4 IBS208P 2 4 35 15 50 Vertebrates(core) 4 IBS209P -

INTRODUCTION to GENETICS Table of Contents Heredity, Historical
INTRODUCTION TO GENETICS Table of Contents Heredity, historical perspectives | The Monk and his peas | Principle of segregation Dihybrid Crosses | Mutations | Genetic Terms | Links Heredity, Historical Perspective | Back to Top For much of human history people were unaware of the scientific details of how babies were conceived and how heredity worked. Clearly they were conceived, and clearly there was some hereditary connection between parents and children, but the mechanisms were not readily apparent. The Greek philosophers had a variety of ideas: Theophrastus proposed that male flowers caused female flowers to ripen; Hippocrates speculated that "seeds" were produced by various body parts and transmitted to offspring at the time of conception, and Aristotle thought that male and female semen mixed at conception. Aeschylus, in 458 BC, proposed the male as the parent, with the female as a "nurse for the young life sown within her". During the 1700s, Dutch microscopist Anton van Leeuwenhoek (1632-1723) discovered "animalcules" in the sperm of humans and other animals. Some scientists speculated they saw a "little man" (homunculus) inside each sperm. These scientists formed a school of thought known as the "spermists". They contended the only contributions of the female to the next generation were the womb in which the homunculus grew, and prenatal influences of the womb. An opposing school of thought, the ovists, believed that the future human was in the egg, and that sperm merely stimulated the growth of the egg. Ovists thought women carried eggs containing boy and girl children, and that the gender of the offspring was determined well before conception. -

Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name GC-rich promoter binding protein 1 Gene Symbol GPBP1 Organism Human Gene Summary This gene was originally isolated by subtractive hybridization of cDNAs expressed in atherosclerotic plaques with a thrombus and was found to be expressed only in vascular smooth muscle cells. However a shorter splice variant was found to be more ubiquitously expressed. This protein is suggested to play a role in the development of atherosclerosis. Studies in mice suggest that it may also function as a GC-rich promoter-specific trans-activating transcription factor. Several alternatively spliced transcript variants encoding different isoforms have been described for this gene. Gene Aliases DKFZp761C169, GPBP, MGC126339, SSH6, VASCULIN RefSeq Accession No. NC_000005.9, NT_006713.15 UniGene ID Hs.444279 Ensembl Gene ID ENSG00000062194 Entrez Gene ID 65056 Assay Information Unique Assay ID qHsaCID0006554 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000424459, ENST00000514387, ENST00000506184, ENST00000511209, ENST00000264779, ENST00000454432, ENST00000538707 Amplicon Context Sequence TGCTCCCTTAACTGAGGATGAAATGAGAGAATTCCAAGTTATTAGTGAACAGTTA CAGAAGAATGGTCTGAGAAAAAATGGTATTTTGAAAAATGGCTTGATCTGTGACT TCAAGTTTGGACCGTGGAAGAACAGCACTTTCAAACCCACAACTGAGAATGATGA CACAGAGACAAGTAG Amplicon Length (bp) 150 Chromosome Location 5:56557058-56558548 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 94 R2 0.9994 cDNA Cq 18.86 Page 1/5 PrimePCR™Assay Validation Report cDNA Tm -

2014-2015 and Is Accurate and Current, to the Greatest Extent Possible, As of June 2014
Cover Cover 1 University’s Mission Statement James B. Duke’s founding Indenture of Duke University directed the members of the University to “provide real leadership in the educational world” by choosing individuals of “outstanding character, ability and vision” to serve as its officers, trustees and faculty; by carefully selecting students of “character, determination and application;” and by pursuing those areas of teaching and scholarship that would “most help to develop our resources, increase our wisdom and promote human happiness.” To these ends, the mission of Duke University is to provide a superior liberal education to undergraduate students, attending not only to their intellectual growth but also to their development as adults committed to high ethical standards and full participation as leaders in their communities; to prepare future members of the learned professions for lives of skilled and ethical service by providing excellent graduate and professional education; to advance the frontiers of knowledge and contribute boldly to the international community of scholarship; to promote an intellectual environment built on a commitment to free and open inquiry; to help those who suffer, cure disease and promote health, through sophisticated medical research and thoughtful patient care; to provide wide ranging educational opportunities, on and beyond our campuses, for traditional students, active professionals and life-long learners using the power of information technologies; and to promote a deep appreciation for the range of human difference and potential, a sense of the obligations and rewards of citizenship, and a commitment to learning, freedom and truth. By pursuing these objectives with vision and integrity, Duke University seeks to engage the mind, elevate the spirit, and stimulate the best effort of all who are associated with the University; to contribute in diverse ways to the local community, the state, the nation and the world; and to attain and maintain a place of real leadership in all that we do. -

Mendelian Gene ” and the ” Molecular Gene ” : Two Relevant Concepts of Genetic Units V Orgogozo, Alexandre E
The ” Mendelian Gene ” and the ” Molecular Gene ” : Two Relevant Concepts of Genetic Units V Orgogozo, Alexandre E. Peluffo, Baptiste Morizot To cite this version: V Orgogozo, Alexandre E. Peluffo, Baptiste Morizot. The ” Mendelian Gene ” and the ”Molec- ular Gene ” : Two Relevant Concepts of Genetic Units. Virginie Orgogozo. Genes and Evolu- tion, 119, Elsevier, pp.1-26, 2016, Current Topics in Developmental Biology, 978-0-12-417194-7. 10.1016/bs.ctdb.2016.03.002. hal-01354346 HAL Id: hal-01354346 https://hal.archives-ouvertes.fr/hal-01354346 Submitted on 19 Aug 2016 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. ARTICLE IN PRESS The “Mendelian Gene” and the “Molecular Gene”: Two Relevant Concepts of Genetic Units V. Orgogozo*,1, A.E. Peluffo*, B. Morizot† *Institut Jacques Monod, UMR 7592, CNRS-Universite´ Paris Diderot, Sorbonne Paris Cite´, Paris, France †Universite´ Aix-Marseille, CNRS UMR 7304, Aix-en-Provence, France 1Corresponding author: e-mail address: [email protected] Contents 1. Introduction 2 2. The “Mendelian Gene” and the “Molecular Gene” 5 3. Current Literature Often Confuses the “Mendelian Gene” and the “Molecular Gene” Concepts 7 4. -

Understanding Multicellular Function and Disease with Human Tissue-Specific Networks
ANALYSIS Understanding multicellular function and disease with human tissue-specific networks Casey S Greene1–3,13, Arjun Krishnan4,13, Aaron K Wong5,13, Emanuela Ricciotti6,7, Rene A Zelaya1, Daniel S Himmelstein8, Ran Zhang9, Boris M Hartmann10, Elena Zaslavsky10, Stuart C Sealfon10, Daniel I Chasman11, Garret A FitzGerald6,7, Kara Dolinski4, Tilo Grosser6,7 & Olga G Troyanskaya4,5,12 Tissue and cell-type identity lie at the core of human tissue- and cell lineage–specific processes1–4. These factors combine physiology and disease. Understanding the genetic to make the understanding of tissue-specific gene functions, disease underpinnings of complex tissues and individual cell lineages is pathophysiology and gene-disease associations particularly challeng- crucial for developing improved diagnostics and therapeutics. ing. Projects such as the Encyclopedia of DNA Elements (ENCODE)5 We present genome-wide functional interaction networks for and The Cancer Genome Atlas (TCGA)6 provide comprehensive 144 human tissues and cell types developed using a data-driven genomic profiles for cell lines and cancers, but the challenge of under- Bayesian methodology that integrates thousands of diverse standing human tissues and cell lineages in the multicellular context of experiments spanning tissue and disease states. Tissue-specific a whole organism remains7. Integrative methods that infer functional networks predict lineage-specific responses to perturbation, gene interaction networks can capture the interplay of pathways, but identify the changing functional roles of genes across tissues existing networks lack tissue specificity8. and illuminate relationships among diseases. We introduce Although direct assay of tissue-specific features remains NetWAS, which combines genes with nominally significant infeasible in many normal human tissues, computational methods genome-wide association study (GWAS) P values and tissue- can infer these features from large data compendia. -

BIO 362 – Introduction to Genetics
PSS6325 – Epigenetic Mechanisms (Fall 2017) 1. Instructor Dr. Benildo G. de los Reyes Professor and Bayer CropScience Endowed Chair in Plant Genomics Department of Plant and Soil Science Experimental Sciences Building, Room 215; 806-834-6421; [email protected] Consultation: By appointment 2. Textbook (Recommended but not required; Available at the library on reserve) Allis CD, Caparros ML, Jenuwein T, Reinberg D (2015) Epigenetics, 2nd Edition, Cold Spring Harbor Laboratory Press, ISBN:978-1-936113-59-0 or newer edition if available. Lecture hand-outs and power point slides will be accessible to students in electronic or other forms. Supplementary printed reading materials will also be provided as necessary. 3. Course description Prerequisites: Consent of instructor; 3-credits, lecture Epigenetics is the study of phenotypic expression that cannot be fully explained by Mendelian principles of heredity, and by DNA sequence variation among individuals. The core of this science is rooted from intrinsically and extrinsically determined processes that lead to dynamic changes in chromatin structure, DNA methylation, RNA- directed gene regulation and genome modification, and how these processes bring about heritable phenotypic variation at the somatic and/or germ levels. During the last decade, the study of epigenetic mechanisms has become a critical aspect in understanding how plants and animals develop and adapt to environmental changes and diseases, providing another layer of complexity to the flow of genetic information as classically defined by the central dogma of molecular biology. This course opens with discussions of the basic concepts and overview of classical examples of epigenetic phenomena in eukaryotes. The initial part of the course will set the framework to examine the various molecular mechanisms that have recently been uncovered in a number of eukaryotic models including yeast, Arabidopsis, maize, C. -

Human Biology - Genetics
Human Biology - Genetics The Program in Human Biology, Stanford University, (HumBio) Say Thanks to the Authors Click http://www.ck12.org/saythanks (No sign in required) www.ck12.org AUTHOR The Program in Human Biology, To access a customizable version of this book, as well as other Stanford University, (HumBio) interactive content, visit www.ck12.org CK-12 Foundation is a non-profit organization with a mission to reduce the cost of textbook materials for the K-12 market both in the U.S. and worldwide. Using an open-content, web-based collaborative model termed the FlexBook®, CK-12 intends to pioneer the generation and distribution of high-quality educational content that will serve both as core text as well as provide an adaptive environment for learning, powered through the FlexBook Platform®. Copyright © 2014 CK-12 Foundation, www.ck12.org The names “CK-12” and “CK12” and associated logos and the terms “FlexBook®” and “FlexBook Platform®” (collectively “CK-12 Marks”) are trademarks and service marks of CK-12 Foundation and are protected by federal, state, and international laws. Any form of reproduction of this book in any format or medium, in whole or in sections must include the referral attribution link http://www.ck12.org/saythanks (placed in a visible location) in addition to the following terms. Except as otherwise noted, all CK-12 Content (including CK-12 Curriculum Material) is made available to Users in accordance with the Creative Commons Attribution-Non-Commercial 3.0 Unported (CC BY-NC 3.0) License (http://creativecommons.org/ licenses/by-nc/3.0/), as amended and updated by Creative Com- mons from time to time (the “CC License”), which is incorporated herein by this reference. -

Genomic Approach in Idiopathic Intellectual Disability Maria De Fátima E Costa Torres
ESTUDOS DE 8 01 PDPGM 2 CICLO Genomic approach in idiopathic intellectual disability Maria de Fátima e Costa Torres D Autor. Maria de Fátima e Costa Torres D.ICBAS 2018 Genomic approach in idiopathic intellectual disability Genomic approach in idiopathic intellectual disability Maria de Fátima e Costa Torres SEDE ADMINISTRATIVA INSTITUTO DE CIÊNCIAS BIOMÉDICAS ABEL SALAZAR FACULDADE DE MEDICINA MARIA DE FÁTIMA E COSTA TORRES GENOMIC APPROACH IN IDIOPATHIC INTELLECTUAL DISABILITY Tese de Candidatura ao grau de Doutor em Patologia e Genética Molecular, submetida ao Instituto de Ciências Biomédicas Abel Salazar da Universidade do Porto Orientadora – Doutora Patrícia Espinheira de Sá Maciel Categoria – Professora Associada Afiliação – Escola de Medicina e Ciências da Saúde da Universidade do Minho Coorientadora – Doutora Maria da Purificação Valenzuela Sampaio Tavares Categoria – Professora Catedrática Afiliação – Faculdade de Medicina Dentária da Universidade do Porto Coorientadora – Doutora Filipa Abreu Gomes de Carvalho Categoria – Professora Auxiliar com Agregação Afiliação – Faculdade de Medicina da Universidade do Porto DECLARAÇÃO Dissertação/Tese Identificação do autor Nome completo _Maria de Fátima e Costa Torres_ N.º de identificação civil _07718822 N.º de estudante __ 198600524___ Email institucional [email protected] OU: [email protected] _ Email alternativo [email protected] _ Tlf/Tlm _918197020_ Ciclo de estudos (Mestrado/Doutoramento) _Patologia e Genética Molecular__ Faculdade/Instituto _Instituto de Ciências