BBS1 Mutations in a Wide Spectrum of Phenotypes Ranging from Nonsyndromic Retinitis Pigmentosa to Bardet-Biedl Syndrome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Educational Paper Ciliopathies
Eur J Pediatr (2012) 171:1285–1300 DOI 10.1007/s00431-011-1553-z REVIEW Educational paper Ciliopathies Carsten Bergmann Received: 11 June 2011 /Accepted: 3 August 2011 /Published online: 7 September 2011 # The Author(s) 2011. This article is published with open access at Springerlink.com Abstract Cilia are antenna-like organelles found on the (NPHP) . Ivemark syndrome . Meckel syndrome (MKS) . surface of most cells. They transduce molecular signals Joubert syndrome (JBTS) . Bardet–Biedl syndrome (BBS) . and facilitate interactions between cells and their Alstrom syndrome . Short-rib polydactyly syndromes . environment. Ciliary dysfunction has been shown to Jeune syndrome (ATD) . Ellis-van Crefeld syndrome (EVC) . underlie a broad range of overlapping, clinically and Sensenbrenner syndrome . Primary ciliary dyskinesia genetically heterogeneous phenotypes, collectively (Kartagener syndrome) . von Hippel-Lindau (VHL) . termed ciliopathies. Literally, all organs can be affected. Tuberous sclerosis (TSC) . Oligogenic inheritance . Modifier. Frequent cilia-related manifestations are (poly)cystic Mutational load kidney disease, retinal degeneration, situs inversus, cardiac defects, polydactyly, other skeletal abnormalities, and defects of the central and peripheral nervous Introduction system, occurring either isolated or as part of syn- dromes. Characterization of ciliopathies and the decisive Defective cellular organelles such as mitochondria, perox- role of primary cilia in signal transduction and cell isomes, and lysosomes are well-known -

Monogenic Causation in Chronic Kidney Disease
University of Dublin, Trinity College School of Medicine, Department of Medicine Investigation of the monogenic causes of chronic kidney disease PhD Thesis April 2020 Dervla Connaughton Supervisor: Professor Mark Little Co-Supervisors: Professor Friedhelm Hildebrandt and Professor Peter Conlon 1 DECLARATION I declare that this thesis has not been submitted as an exercise for a degree at this or any other university and it is entirely my own work. This work was funding by the Health Research Board, Ireland (HPF-206-674), the International Pediatric Research Foundation Early Investigators’ Exchange Program and the Amgen® Irish Nephrology Society Specialist Registrar Bursary. I agree to deposit this thesis in the University’s open access institutional repository or allow the Library to do so on my behalf, subject to Irish Copyright Legislation and Trinity College Library conditions of use and acknowledgement. I consent to the examiner retaining a copy of the thesis beyond the examining period, should they so wish (EU GDPR May 2018). _____________________ Dervla Connaughton 2 SUMMARY Chapter 1 provides an introduction to the topic while Chapter 2 provides details of the methods employed in this work. In Chapter 3 I provide an overview of the currently known monogenic causes for human chronic kidney disease (CKD). I also describe how next- generation sequencing can facilitate molecular genetic diagnostics in individuals with suspected genetic kidney disease. Chapter 4 details the findings of a multi-centre, cross-sectional study of patients with CKD in the Republic of Ireland. The primary aim of this study (the Irish Kidney Gene Project) was to describe the prevalence of reporting a positive family history of CKD among a representation sample of the CKD population. -

Diagnostic Test: OBESITÀ GENETICHE MENDELIANE
Diagnostic test: OBESITÀ GENETICHE MENDELIANE MENDELIAN OBESITY Panel / Illumina Custom panel, Nextera Enrichment Technology / Coding exons and flanking regions of genes List of gene(s) and disease(s) tested: ALMS1, ARL6, BBIP1, BBS1, BBS10, BBS12, BBS2, BBS4, BBS5, BBS7, BBS9, C8orf37, CARTPT, CEP19, CEP290, DYRK1B, GNAS, HDAC8, IFT172, IFT27, INPP5E, INSR, KSR2, LEP, LEPR, LZTFL1, MC3R, MC4R, MCHR1, MEGF8, MKKS, MKS1, NR0B2, PCSK1, PHF6, POMC, PPARG, PPP1R3A, RAB23, SDCCAG8, SH2B1, SIM1, TRIM32, TTC8, UCP3, VPS13B, WDPCP ORPHA:98267 Obesità non sindromica genetica Obesità sindromica Tabella Elenco delle forme di OBESITÀ GENETICHE MENDELIANE e la loro eziologia genetica Phenotype OMIM# Gene OMIM# Phenotype Gene Alstrom syndrome 203800 ALMS1 606844 Bardet-Biedl syndrome 3 600151 ARL6 608845 Bardet-Biedl syndrome 18 615995 BBIP1 613605 Bardet-Biedl syndrome 1 209900 BBS1 209901 Bardet-Biedl syndrome 10 615987 BBS10 610148 Bardet-Biedl syndrome 12 615989 BBS12 610683 Bardet-Biedl syndrome 2 615981 BBS2 606151 Bardet-Biedl syndrome 4 615982 BBS4 600374 Bardet-Biedl syndrome 5 615983 BBS5 603650 Bardet-Biedl syndrome 7 615984 BBS7 607590 Bardet-Biedl syndrome 21 617406 C8orf37 614477 Obesity, severe HGMD CARTPT 602606 Morbid obesity and spermatogenic failure; Bardet-Biedl syndrome; Morbid obesity 615703; HGMD CEP19 615586 Bardet-Biedl syndrome 14 615991 CEP290 610142 Abdominal obesity-metabolic syndrome 3 615812 DYRK1B 604556 Pseudohypoparathyroidism Ia; Pseudohypoparathyroidism Ic 103580; 612462 GNAS 139320 Cornelia de Lange syndrome 5 300882 -

Renal Cystic Disorders Infosheet 6-14-19
Next Generation Sequencing Panel for Renal Cystic Disorders Clinical Features: Renal cystic diseases are a genetically heterogeneous group of conditions characterized By isolated renal disease or renal cysts in conjunction with extrarenal features (1). Age of onset of renal cystic disease ranges from neonatal to adult onset. Common features of renal cystic diseases include renal insufficiency and progression to end stage renal disease (ESRD). Identification of the genetic etiology of renal cystic disease can aid in appropriate clinical management of the affected patient. Our Renal Cystic Disorders Panel includes sequence and deletion/duplicaton analysis of all 79 genes listed below. Renal Cystic Disorders Sequencing Panel AHI1 BMPER HNF1B NEK8 TCTN3 WDPCP ANKS6 C5orf42 IFT27 NOTCH2 TFAP2A WDR19 ARL13B CC2D2A IFT140 NPHP1 TMEM107 XPNPEP3 ARL6 CDC73 IFT172 NPHP3 TMEM138 ZNF423 B9D1 CEP104 INPP5E NPHP4 TMEM216 B9D2 CEP120 INVS OFD1 TMEM231 BBIP1 CEP164 IQCB1 PDE6D TMEM237 BBS1 CEP290 JAG1 PKD2 TMEM67 BBS10 CEP41 KIAA0556 PKHD1 TRIM32 BBS12 CEP83 KIAA0586 REN TSC1 BBS2 CRB2 KIF14 RPGRIP1L TSC2 BBS4 CSPP1 KIF7 SALL1 TTC21B BBS5 DCDC2 LZTFL1 SDCCAG8 TTC8 BBS7 GLIS2 MKKS TCTN1 UMOD BBS9 GLIS3 MKS1 TCTN2 VHL Disorder Genes Inheritance Clinical features/molecular genetics Bardet Biedl ARL6 AR Bardet-Biedl syndrome (BBS) is an autosomal syndrome BBS1 recessive multi-systemic ciliopathy characterized By BBS10 retinal dystrophy, oBesity, postaxial polydactyly, BBS12 leaning difficulties, renal involvement and BBS2 genitourinary abnormalities (2). Visual prognosis is BBS4 poor, and the mean age of legal Blindness is 15.5 BBS5 years. Birth weight is typically normal But significant BBS7 weight gain Begins within the first year. Renal BBS9 disease is a major cause of morBidity and mortality. -

Modeling Ciliopathies: Primary Cilia in Development and Disease
CHAPTER FIVE Modeling Ciliopathies: Primary Cilia in Development and Disease Robyn J. Quinlan, Jonathan L. Tobin, and Philip L. Beales Contents 1. The Human Ciliopathies 250 2. Bardet-Biedl Syndrome 251 3. Alstro¨m Syndrome 263 4. Polycystic Kidney Disease 264 5. Nephronophthisis 265 6. Meckel Syndrome 267 7. Joubert Syndrome 268 8. Jeune Syndrome 269 9. Oral–Facial–Digital Syndrome 269 10. The Structure and Function of the Cilium 270 10.1. The structure of primary cilia 270 10.2. Intraflagellar transport 272 10.3. IFT and ciliopathies 272 11. Cilia and Development 276 11.1. Left–right determination 276 11.2. Hedgehog signaling in ciliopathies 277 11.3. Cilia and polarity 280 11.4. Cilia and cystogenesis in mammals 283 11.5. Cilia and obesity 286 12. Therapies for Cystic Disease 287 13. Concluding Remarks 289 References 292 Abstract Primary (nonmotile) cilia are currently enjoying a renaissance in light of novel ascribed functions ranging from mechanosensory to signal transduction. Their importance for key developmental pathways such as Sonic Hedgehog (Shh) and Wnt is beginning to emerge. The function of nodal cilia, for example, is vital for Molecular Medicine Unit, Institute of Child Health, University College London, London, WC1N1EH, United Kingdom Current Topics in Developmental Biology, Volume 84 # 2008 Elsevier Inc. ISSN 0070-2153, DOI: 10.1016/S0070-2153(08)00605-4 All rights reserved. 249 250 Robyn J. Quinlan et al. breaking early embryonic symmetry, Shh signaling is important for tissue morphogenesis and successful Wnt signaling for organ growth and differentia- tion. When ciliary function is perturbed, photoreceptors may die, kidney tubules develop cysts, limb digits multiply and brains form improperly. -

Evolution of Modular Intraflagellar Transport from a Coatomer-Like
Evolution of modular intraflagellar transport from a coatomer-like progenitor Teunis J. P. van Dama, Matthew J. Townsendb, Martin Turkc,1, Avner Schlessingerc,2, Andrej Salic,d,e, Mark C. Fieldb,3, and Martijn A. Huynena,3,4 aCentre for Molecular and Biomolecular Informatics, Radboud University Medical Centre, 6500 HB, Nijmegen, The Netherlands; bDepartment of Pathology, University of Cambridge, Cambridge CB2 1QP, United Kingdom; and cDepartment of Bioengineering and Therapeutic Sciences, dDepartment of Pharmaceutical Chemistry, and eCalifornia Institute for Quantitative Biosciences, University of California, San Francisco, CA 94158 Edited by Russell F. Doolittle, University of California at San Diego, La Jolla, CA, and approved March 15, 2013 (received for review December 4, 2012) The intraflagellar transport (IFT) complex is an integral component NPC scaffold (2–4). This classification was based on sequence of the cilium, a quintessential organelle of the eukaryotic cell. The similarity of IFT subunits to the COPI-α and -β′ subunits, further IFT system consists of three subcomplexes [i.e., intraflagellar supported by secondary structure predictions. However, a full transport (IFT)-A, IFT-B, and the BBSome], which together trans- phylogenetic reconstruction and structural analysis of the IFT port proteins and other molecules along the cilium. IFT dysfunction complex has not been performed. Such an analysis is necessary results in diseases collectively called ciliopathies. It has been pro- because the abundance of the WD40 and TPR domains in non- posed that the IFT complexes originated from vesicle coats similar coatomer subunit proteins requires more than sequence similarity to coat protein complex (COP) I, COPII, and clathrin. Here we to establish a close phylogenetic relationship. -
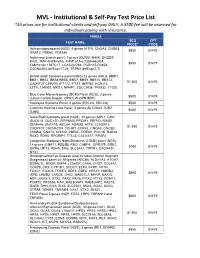
MVL - Institutional & Self-Pay Test Price List *All Prices Are for Institutional Clients and Self-Pay ONLY
MVL - Institutional & Self-Pay Test Price List *All prices are for institutional clients and self-pay ONLY. A $500 fee will be assessed for individuals paying with insurance. PANELS SEQ CPT TEST NAME PRICE* CODE Achromatopsiapanel (NGS), 6 genes (ATF6, CNGA3, CNGB3, $950 81479 GNAT2, PDE6C, PDE6H) Ashkenazi Jewish panel, 7 genes (CLRN1-N48K, DHDDS- K42E, MAK-K429insAlu, FAM161A-c.1355-6delCA, $550 81407 FAM161Ac.1567C>T, LCA5-Q279X, PCDH15-R245X, CACNA2D4-delExon17-26, TRPM1-delExon2-7) Bardet-Biedl Syndrome panel (NGS) 23 genes (ARL6, BBIP1, BBS1, BBS2, BBS4,BBS5, BBS7, BBS9, BBS10, BBS12, $1,500 81479 C8ORF37,CEP290, IFT172, IFT27, INPP5E, KCNJ13, LZTFL1,MKKS, MKS1, NPHP1, SDCCAG8, TRIM32, TTC8) Blue Cone Monochromacy (BCM) Panel (NGS), 2 genes $500 81479 (Locus Control Region, OPN1LW-OPN1MW) Cockayne Sydrome Panel, 2 genes (ERCC6, ERCC8) $500 81479 Common Hearing Loss Panel, 3 genes (SLC26A4, GJB2, $650 81479 GJB6) Cone-Rod Dystrophy panel (NGS), 33 genes (AIPL1, CRX, GUCA1A, GUCY2D, PITPNM3, PROM1, PRPH2, RIMS1, SEMA4A, UNC119, ABCA4, ADAM9, ATF6, C21ORF2, $1,950 81479 C8ORF37,CACNA2D4, CDHR1, CERKL, CNGA3, CNGB3, CNNM4, GNAT2, KCNV2, PDE6C, PDE6H, POC1B, RAB28, RAX2, RDH5, RPGRIP1, TTLL5, CACNA1F, RPGR) Congenital Stationary Night Blindness (CSNB) panel (NGS), 14 genes (GNAT1, PDE6B, RHO, CABP4, GPR179, GRK1, $950 81479 GRM6,LRIT3, RDH5, SAG, SLC24A1, TRPM1, CACNA1F, NYX) Developmental Eye Disease (also includes Anterior Segment Dysgenesis) panel v4: 59 genes (ABCB6, ALDH1A3, ATOH7, B3GALTL, BCOR, BMP4, c12orf57, CASK, CHD7, COL4A1, -

Targeted High-Throughput Sequencing for Diagnosis of Genetically Heterogeneous Diseases: Efficient Mutation Detection in Bardet
Methods J Med Genet: first published as 10.1136/jmedgenet-2012-100875 on 7 July 2012. Downloaded from ORIGINAL ARTICLE Targeted high-throughput sequencing for diagnosis of genetically heterogeneous diseases: efficient mutation detection in Bardet-Biedl and Alstro¨m Syndromes Claire Redin,1 Ste´phanie Le Gras,2 Oussema Mhamdi,3 Ve´ronique Geoffroy,4 Corinne Stoetzel,5 Marie-Claire Vincent,6 Pietro Chiurazzi,7 Didier Lacombe,8 Ines Ouertani,3 Florence Petit,9 Marianne Till,10 Alain Verloes,11 Bernard Jost,2 Habiba Bouhamed Chaabouni,3 Helene Dollfus,5,12 Jean-Louis Mandel,1,6,13 Jean Muller1,6 < Additional materials are ABSTRACT INTRODUCTION published online only. To view Background Bardet-Biedl syndrome (BBS) is Bardet-Biedl syndrome (BBS; OMIM# 209900) is these files please visit the a pleiotropic recessive disorder that belongs to the a pleiotropic recessive disorder with high non-allelic journal online (http://dx.doi.org/ 10.1136/jmedgenet-2012- rapidly growing family of ciliopathies. It shares genetic heterogeneity. Its incidence varies from an 100875/content/early/recent). phenotypic traits with other ciliopathies, such as Alstro¨m estimated 1:160 000 in northern Europe to e For numbered affiliations see syndrome (ALMS), nephronophthisis (NPHP) or Joubert 1:13 500 17 000 in Bedouins and Newfound- 1 end of article. syndrome. BBS mutations have been detected in 16 landers, respectively. BBS belongs to the large and different genes (BBS1-BBS16) without clear genotype- growing family of ciliopathies and, therefore, shares Correspondence to to-phenotype correlation. This extensive genetic phenotypic traits with Joubert (JBTS), Alström Professor Jean-Louis Mandel, heterogeneity is a major concern for molecular diagnosis (ALMS) and Meckel (MKS) syndromes.12Differ- Department of Neurogenetics & fi Translational medicine, IGBMC, and genetic counselling. -

Ciliopathies: Genetics in Pediatric Medicine
Review Article Ciliopathies: Genetics in Pediatric Medicine Machteld M. Oud1 IdekeJ.C.Lamers1 Heleen H. Arts1,2 1 Department of Human Genetics, Radboud Institute for Molecular Life Address for correspondence Heleen H. Arts, Department of Human Sciences, Radboud University Medical Center, Nijmegen, Genetics, Radboud Institute for Molecular Life Sciences, Radboud The Netherlands University Medical Center, PO-Box 9101, 6500 HB, Nijmegen, 2 Department of Biochemistry, University of Western Ontario, London, The Netherlands (e-mail: [email protected]). Ontario, Canada JPediatrGenet Abstract Ciliary disorders,whicharealsoreferredtoasciliopathies, are a group of hereditary disorders that result from dysfunctional cilia. The latter are cellular organelles that stick up from the apical plasma membrane. Cilia have important roles in signal transduction and facilitate communications between cells and their surroundings. Ciliary disruption can result in a wide variety of clinically and genetically heterogeneous disorders with overlapping phenotypes. Because cilia occur widespread in our bodies many organs and sensory systems can be affected when they are dysfunctional. Ciliary disorders may be isolated or syndromic, and common features are cystic liver and/or kidney disease, blindness, neural tube defects, brain anomalies and intellectual disability, skeletal abnormalities ranging from polydactyly to abnormally short ribs and limbs, ectodermal defects, obesity, situs inversus, infertility, and recurrent respiratory tract infections. In this -

Mouse Models of Inherited Retinal Degeneration with Photoreceptor Cell Loss
cells Review Mouse Models of Inherited Retinal Degeneration with Photoreceptor Cell Loss 1, 1, 1 1,2,3 1 Gayle B. Collin y, Navdeep Gogna y, Bo Chang , Nattaya Damkham , Jai Pinkney , Lillian F. Hyde 1, Lisa Stone 1 , Jürgen K. Naggert 1 , Patsy M. Nishina 1,* and Mark P. Krebs 1,* 1 The Jackson Laboratory, Bar Harbor, Maine, ME 04609, USA; [email protected] (G.B.C.); [email protected] (N.G.); [email protected] (B.C.); [email protected] (N.D.); [email protected] (J.P.); [email protected] (L.F.H.); [email protected] (L.S.); [email protected] (J.K.N.) 2 Department of Immunology, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok 10700, Thailand 3 Siriraj Center of Excellence for Stem Cell Research, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok 10700, Thailand * Correspondence: [email protected] (P.M.N.); [email protected] (M.P.K.); Tel.: +1-207-2886-383 (P.M.N.); +1-207-2886-000 (M.P.K.) These authors contributed equally to this work. y Received: 29 February 2020; Accepted: 7 April 2020; Published: 10 April 2020 Abstract: Inherited retinal degeneration (RD) leads to the impairment or loss of vision in millions of individuals worldwide, most frequently due to the loss of photoreceptor (PR) cells. Animal models, particularly the laboratory mouse, have been used to understand the pathogenic mechanisms that underlie PR cell loss and to explore therapies that may prevent, delay, or reverse RD. Here, we reviewed entries in the Mouse Genome Informatics and PubMed databases to compile a comprehensive list of monogenic mouse models in which PR cell loss is demonstrated. -

What's New in Genetics
Tuesday, 10:00 – 11:30, A1 What's New in Genetics Helga V. Toriello [email protected] Objective: Identify advances in clinical assessment and management of selected healthcare issues related to persons with developmental disabilities Notes: 4/10/2017 PRESENTATION TOPICS What’s New in • THE ISSUE OF THE UNDIAGNOSED Genetics PATIENT • TESTING FOR GENETIC CONDITIONS Helga V. Toriello, PhD • TREATMENT OPTIONS FOR THE FUTURE UNDIAGNOSED PATIENT UNDIAGNOSED PATIENT • Genetic disorders are individually rare, but • Undiagnosed diseases remain undiagnosed if cumulatively affect 25 million in the US alone incorrectly diagnosed as something else. example. • Harder for physicians to recognize most of these rare • Problem if wrong diagnosis persists – wrong disorders; initial findings non-specific, e.g., weight treatment, wrong natural history, evaluations which loss, growth failure, fatigue, fever are unnecessary are done and vice versa • Tendency is to make diagnosis of common disorder • Clinic experience with the “why isn’t this child rather than rare disorder deceased yet?” referrals EXAMPLES DIAGNOSTIC PITFALLS Initial diagnosis Final diagnosis Late-onset autistic regression Kleefstra syndrome • Inherent to disease process Hypotonia Nemaline myopathy • Seizures SCN2B mutation Patient-specific IBD XIAP • Physician-specific Arthritis Hyper-IgD syndrome • Limitations in diagnostic modalities Hepatic failure Citrin deficiency 1 4/10/2017 DISEASE PROCESS PATIENT SPECIFIC • “diseases do not read the textbooks” • Parents often anxious, -

Ciliopathies
Ciliopathies About Emory Genetics Laboratory (EGL) EGL specializes in genetic diagnostic testing, with 45 years of clinical experience and board-certified laboratory directors and genetic counselors reporting out cases. EGL offers a combined 1100 molecular genetics, biochemical genetics, and cytogenetics tests under one roof and custom testing for all medically relevant genes, for domestic and international clients. Equally important to improving patient care through quality genetic testing is the contribution EGL makes back to the scientific and medical communities. EGL is one of only a few clinical diagnostic laboratories to openly share data with the NCBI freely available public database ClinVar (>7000 variants on >500 genes) and is also the only laboratory with a free online database (EmVClass), featuring a variant classification search and report request interface, which facilitates rapid interactive curation and reporting of variants. Ciliopathies Panel The ciliopathies are a group of disorders caused by mutation in genes that encode proteins involved in the formation and function of cilia. Cilia are microtubule-based, hair-like cytoplasmic extensions from the cell surface and are found on almost all cell types. They play a part in multiple biological processes, including cellular motility, extracellular fluid movement, and sensory and signal transduction pathways. As many developmental and physiological functions are dependent on cilia, defects in cilia may lead to disorders in many different organ systems and processes, including: • renal disease • cerebral anomalies • congenital fibrocystic diseases • retinal degeneration • skeletal dysplasia of the pancreas and liver • diabetes • obesity There are 112 genes on the Ciliopathies Panel (please see reverse side for a list of genes analyzed).