Meier-Gorlin Syndrome Sonja A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Neonatal Orthopaedics
NEONATAL ORTHOPAEDICS NEONATAL ORTHOPAEDICS Second Edition N De Mazumder MBBS MS Ex-Professor and Head Department of Orthopaedics Ramakrishna Mission Seva Pratishthan Vivekananda Institute of Medical Sciences Kolkata, West Bengal, India Visiting Surgeon Department of Orthopaedics Chittaranjan Sishu Sadan Kolkata, West Bengal, India Ex-President West Bengal Orthopaedic Association (A Chapter of Indian Orthopaedic Association) Kolkata, West Bengal, India Consultant Orthopaedic Surgeon Park Children’s Centre Kolkata, West Bengal, India Foreword AK Das ® JAYPEE BROTHERS MEDICAL PUBLISHERS (P) LTD. New Delhi • London • Philadelphia • Panama (021)66485438 66485457 www.ketabpezeshki.com ® Jaypee Brothers Medical Publishers (P) Ltd. Headquarters Jaypee Brothers Medical Publishers (P) Ltd. 4838/24, Ansari Road, Daryaganj New Delhi 110 002, India Phone: +91-11-43574357 Fax: +91-11-43574314 Email: [email protected] Overseas Offices J.P. Medical Ltd. Jaypee-Highlights Medical Publishers Inc. Jaypee Brothers Medical Publishers Ltd. 83, Victoria Street, London City of Knowledge, Bld. 237, Clayton The Bourse SW1H 0HW (UK) Panama City, Panama 111, South Independence Mall East Phone: +44-2031708910 Phone: +507-301-0496 Suite 835, Philadelphia, PA 19106, USA Fax: +02-03-0086180 Fax: +507-301-0499 Phone: +267-519-9789 Email: [email protected] Email: [email protected] Email: [email protected] Jaypee Brothers Medical Publishers (P) Ltd. Jaypee Brothers Medical Publishers (P) Ltd. 17/1-B, Babar Road, Block-B, Shaymali Shorakhute, Kathmandu Mohammadpur, Dhaka-1207 Nepal Bangladesh Phone: +00977-9841528578 Mobile: +08801912003485 Email: [email protected] Email: [email protected] Website: www.jaypeebrothers.com Website: www.jaypeedigital.com © 2013, Jaypee Brothers Medical Publishers All rights reserved. No part of this book may be reproduced in any form or by any means without the prior permission of the publisher. -

Basic Biomechanics
Posture analysis • A quick evaluation of structure and function • Doctor views patient from behind (P-A) and from the side (lateral) • References points of patient’s anatomy relative to plumb (a position of balance) 9/3/2013 1 Posture analysis • Lateral View – Knees (anterior, posterior, plumb, genu recurvatum) – Trochanter (anterior, posterior, plumb) – Pelvis (anterior, posterior, neutral pelvic tilt) – Lumbar lordosis (hypo-, hyper-, normal) – Mid-axillary line (anterior, posterior, plumb) – Thoracic kyphosis (hyp-, hyper- normal) – Acromion (anterior, posterior, plumb) – Scapulae (protracted, retracted, normal) – Cervical lordosis (hypo-, hyper-, normal) – External auditory meatus (anterior, posterior, plumb) – Occiput (extended, neutral, flexed) 9/3/2013 2 Posture analysis • Posterior – Anterior View – Feet (pronation, supination, normal) – Achilles tendon (bowed in/out, normal) – Knees (genu valga/vera, normal - internal/external rotation) – Popliteal crease heights (low, high, level) – Trochanter heights (low, high, level) – Iliac crest heights (low on the right/left, normal) – Lumbar scoliosis (right/left, or no signs of) – Thoracic scoliosis (right/left, or no signs of) – Shoulder level (low on the right/left, or normal) – Cervical scoliosis (right/left, or no signs of) – Cervical position (rotation, tilt, neutral) – Mastoid (low on the right/left, or normal) 9/3/2013 3 …..poor postures 9/3/2013 4 Functional Anatomy of the Spine • The vertebral curvatures – Cervical Curve • Anterior convex curve (lordosis) develop in infancy -

ICD-9CM Coding Achilles Bursitis Or Tendinitis 726.71 Adhesive
ICD-9CM CODING OF COMMON CHIROPRACTIC CONDITIONS CONDITION ICD-9CM coding Achilles bursitis or tendinitis 726.71 Adhesive capsulitis of shoulder 726 Anklyosing Spondylitis (spine only) 720 Anterior cruciate ligament (old disruption) 717.83 Bicipital tenosynovitis 726.12 Boutonniere deformity 736.21 Brachial neuritis or radiculitis 723.4 Bunion 727.1 Bursitis of knee 726.6 Calcaneal spur 726.73 Carpal tunnel syndrome 354 Cervical radiculitis 723.4 Cervical spondylosis with myelopathy 721.1 Cervical spondylosis without myelopathy 721 Cervicalgia 723.1 Chondromalacia of patella 717.7 Claw hand (acquired) 736.06 Claw toe (acquired) 735.5 Coccygodynia 724.79 Coxa plana 732.1 Coxa valga (acquired) 736.31 Coxa vara (acquired) 736.32 de Quervain's disease 727.04 Degeneration of cervical intervertebral disc 722.4 Degeneration of thoracic or lumbar intervertebral disc 722.5 Disc Degeneration (Cervical with myelopathy) 722.71 Disc Degeneration (Lumbar with myelopathy) 722.73 Disc Degeneration (Thoracic) 722.51 Disc Degeneration (Cervical) 722.4 Disc Degeneration (Lumbar) 722.52 Disc Degeneration (Thoracic with myelopathy) 722.72 Disc displacement without myelopathy (Thoracic) 722.11 Disc displacement of without myelopathy (Cervical ) 722 Disc displacement without myelopathy (Lumbar) 722.1 Dupuytren's contracture 728.6 Entrapment syndromes 354.0-355.9 Epicondylitis (Lateral) 726.32 Epicondylitis (Medial) 726.31 Flat foot-Pes planus (acquired) 734 Fracture (Lumbar) 805.4 Ganglion of tendon sheath 727.42 Genu recurvatum (acquired) 736.5 Genu valgum -

Posture Analysis
POSTURE ANALYSIS PRESENTED BY MWADZIWANA LOUIS LAW WHAT IS POSTURE ? • Posture is a position of greatest efficiency, around your center of gravity, with muscles on all sides, exerting pull equally. CORRECT POSTURE Correct posture “Position in which minimum stress is placed on each joint.” (Magee) Maintains the natural curves Faulty posture Any position that increases stress on joints Create muscle imbalances, ligamentous tension, circulatory occlusion CAUSES OF POOR POSTURE • Positional factors/Habitual • Appearance of increased height (social stigma) • Muscle imbalances/contractures • Pain e.g. ICD pleural effusion • Respiratory conditions CAUSES OF POOR POSTURE • Structural factors • Congenital anomalies • Developmental problems • Trauma • Disease FACTORS AFFECTING POSTURAL ANALYSIS • Subject must be minimally clothed • The subject must assume a comfortable and relaxed posture • Subjects who use orthotic or assistive devices should be assessed with and without them to determine their effectiveness in correcting posture. • relevant medical history and other information THE SPINAL COLUMN • Primary curves • Thoracic spine • Sacrum • Secondary curves • Cervical spine • Lumbar spine LATERAL VIEW • Head and neck: • Plumb line: The line falls through the ear lobe to the acromion process. • Common faults include: • Forward head: • Flattened lordotic cervical curve • Excessive Lordotic curve LATERAL VIEW Shoulder: • Plumb line: It falls through the acromion process. • Common faults include: • Forward shoulders • Lumbar Lordosis LATERAL VIEW • -

Osteotomy Around the Knee: Evolution, Principles and Results
Knee Surg Sports Traumatol Arthrosc DOI 10.1007/s00167-012-2206-0 KNEE Osteotomy around the knee: evolution, principles and results J. O. Smith • A. J. Wilson • N. P. Thomas Received: 8 June 2012 / Accepted: 3 September 2012 Ó Springer-Verlag 2012 Abstract to other complex joint surface and meniscal cartilage Purpose This article summarises the history and evolu- surgery. tion of osteotomy around the knee, examining the changes Level of evidence V. in principles, operative technique and results over three distinct periods: Historical (pre 1940), Modern Early Years Keywords Tibia Osteotomy Knee Evolution Á Á Á Á (1940–2000) and Modern Later Years (2000–Present). We History Results Principles Á Á aim to place the technique in historical context and to demonstrate its evolution into a validated procedure with beneficial outcomes whose use can be justified for specific Introduction indications. Materials and methods A thorough literature review was The concept of osteotomy for the treatment of limb defor- performed to identify the important steps in the develop- mity has been in existence for more than 2,000 years, and ment of osteotomy around the knee. more recently pain has become an additional indication. Results The indications and surgical technique for knee The basic principle of osteotomy (osteo = bone, tomy = osteotomy have never been standardised, and historically, cut) is to induce a surgical transection of a bone to allow the results were unpredictable and at times poor. These realignment and a consequent transfer of weight bearing factors, combined with the success of knee arthroplasty from a damaged area to an undamaged area of joint surface. -

Page 1 of 4 COPYRIGHT © by the JOURNAL of BONE and JOINT SURGERY, INCORPORATED LAMPLOT ET AL
COPYRIGHT © BY THE JOURNAL OF BONE AND JOINT SURGERY, INCORPORATED LAMPLOT ET AL. RISK OF SUBSEQUENT JOINT ARTHROPLASTY IN CONTRALATERAL OR DIFFERENT JOINT AFTER INDEX SHOULDER, HIP, OR KNEE ARTHROPLASTY http://dx.doi.org/10.2106/JBJS.17.00948 Page 1 Appendix TABLE E-1 Included Alternative Primary Diagnoses ICD-9-CM Code Diagnosis* 716.91 Arthropathy NOS, shoulder 716.95 Arthropathy NOS, pelvis 716.96 Arthropathy NOS, lower leg 719.45 Joint pain, pelvis 719.91 Joint disease NOS, shoulder *NOS = not otherwise specified. Page 1 of 4 COPYRIGHT © BY THE JOURNAL OF BONE AND JOINT SURGERY, INCORPORATED LAMPLOT ET AL. RISK OF SUBSEQUENT JOINT ARTHROPLASTY IN CONTRALATERAL OR DIFFERENT JOINT AFTER INDEX SHOULDER, HIP, OR KNEE ARTHROPLASTY http://dx.doi.org/10.2106/JBJS.17.00948 Page 2 TABLE E-2 Excluded Diagnoses* ICD-9- ICD-9- ICD-9- ICD-9- CM Code Diagnosis CM Code Diagnosis CM Code Diagnosis CM Code Diagnosis 274 Gouty arthropathy NOS 696 Psoriatic 711.03 Pyogen 711.38 Dysenter arthropathy arthritis- arthritis NEC forearm 274.01 Acute gouty arthropathy 696.1 Other psoriasis 711.04 Pyogen 711.4 Bact arthritis- arthritis-hand unspec 274.02 Chr gouty arthropathy 696.2 Parapsoriasis 711.05 Pyogen 711.46 Bact arthritis- w/o tophi arthritis-pelvis l/leg 274.03 Chr gouty arthropathy w 696.3 Pityriasis rosea 711.06 Pyogen 711.5 Viral arthritis- tophi arthritis-l/leg unspec 274.1 Gouty nephropathy NOS 696.4 Pityriasis rubra 711.07 Pyogen 711.55 Viral arthritis- pilaris arthritis-ankle pelvis 274.11 Uric acid nephrolithiasis 696.5 Pityriasis NEC & 711.08 -

Congenital Anomalies and in Utero Antiretroviral Exposure in Human Immunodeficiency Virus– Exposed Uninfected Infants
Supplementary Online Content Williams PL, Crain MJ, Yildirim C, et al; Pediatric HIV/AIDS Cohort Study. Congenital anomalies and in utero antiretroviral exposure in human immunodeficiency virus– exposed uninfected infants. Published online November 10, 2014. JAMA Pediatr. doi:10.1001/jamapediatrics.2014.1889. eTable 1. Frequency of Specific Major Congenital Anomalies Within Anomaly Categories eTable 2. Anomalies Reported Among Children Exposed to Atazanavir During the First Trimester eTable 3. Association of Timing of the First ARV Exposure During Pregnancy With Congenital Anomalies by ARV Drug Class and for Specific ARV Drugs This supplementary material has been provided by the authors to give readers additional information about their work. © 2014 American Medical Association. All rights reserved. Downloaded From: https://jamanetwork.com/ on 09/24/2021 eTable 1. Frequency of Specific Major Congenital Anomalies Within Anomaly Categories # of children with at Total # of least one major anomaly in Anomaly anomalies category Category (Total=242) (Total=201) List of Major Anomalies Musculoskeletal 72 59 Polydactyly (15), torticollis/muscular anomaly (12), clubfoot, talipes, other foot deformity (9), congenital dislocation of hip (5), craniosynostosis (4), plagiocephaly (4), pectus excavatum/funnel chest (3), lower limb anomaly (3), hypertelorism/other face or skull anomaly (3), spina bifida occulta/spine anomaly (3), syndactyly (3), inguinal hernia (2), diaphragmatic hernia/Morgagni, genu recurvatum/bowed legs, rib/sternum anomaly, scoliosis/congenital -

Equinus Deformity in the Pediatric Patient: Causes, Evaluation, and Management
Equinus Deformity in the Pediatric Patient: Causes, Evaluation, and Management a,b,c Monique C. Gourdine-Shaw, DPM, LCDR, MSC, USN , c, c Bradley M. Lamm, DPM *, John E. Herzenberg, MD, FRCSC , d,e Anil Bhave, PT KEYWORDS Equinus Pediatric External fixation Achilles tendon lengthening Gastrocnemius recession Tendo-Achillis lengthening Different body and limb segments grow at different rates, inducing varying muscle tensions during growth.1 In addition, boys and girls grow at different rates.1 The rate of growth for girls spikes at ages 5, 7, 10, and 13 years.1 The estrogen-induced pubertal growth spurt in girls is one of the earliest manifestations of puberty. Growth of the legs and feet accelerates first, so that many girls have longer legs in proportion to their torso during the first year of puberty. The overall rate of growth tends to reach a peak velocity (as much as 7.5 to 10 cm) midway between thelarche and menarche and declines by the time menarche occurs.1 In the 2 years after menarche, most girls grow approximately 5 cm before growth ceases at maximal adult height.1 The rate of growth for boys spikes at ages 6, 11, and 14 years.1 Compared with girls’ early growth spurt, growth accelerates more slowly in boys and lasts longer, resulting in taller adult stature among men than women (on average, approximately 10 cm).1 The difference is attributed to the much greater potency of estradiol compared with testosterone in Two authors (BML and JEH) host an international teaching conference supported by Smith & Nephew. -

Pediatric Flatfoot Deformity Case Study Evaluation and Management Frank a Luckino III DPM, Jeffrey C
Pediatric Flatfoot Deformity Case Study Evaluation and Management Frank A Luckino III DPM, Jeffrey C. Lupica DPM, Allan Boike DPM Department of Podiatry Pediatric flatfoot is not an uncommon patient presentation in clinical practice. A thorough history and physical examination of these children is Treatment of pediatric pes planus depends on the degree of deformity and Parents often become more concerned than the children themselves. Reports paramount. Standing exam and gait analysis is recommended. Body symptomatology. Initial treatment should consist of modifying activity and non- show that more than 30% of all newborns have a calcaneal valgus foot habitus and limb alignment should be noted as well (1). Radiographs in a steroidal anti-inflammatory medications as necessary (2). Stretching regimens deformity of both feet (5). As these individuals mature, the majority will weight-bearing fashion should be obtained (5). As a clinician, one needs to may be implemented if an equinus deformity is a cause for abnormal pronation become asymptomatic and not necessitate care. However, some patients will determine whether the deformity is pathologic or non-pathologic, rigid or and subsequent pes planus. Physical therapy should be started when muscle require conservative and or surgical treatment. The goal of the clinician is flexible, functional or nonfunctional, symptomatic or asymptomatic (1). weakness is observed. Orthotics, whether over-the counter or custom made, determine whether the deformity is rigid or flexible, painful or non-painful, and A calcaneovalgus foot deformity presents with a flexible, deformity which is may provide some relief despite limited level one studies (1). In an article by functional versus nonfunctional. -

Report of a Sporadic Caudal Regression Syndrome with Cleft Lip
Running title : Caudal Regression Syndrome with CLP Nitte University Journal of Health Science Case Report Report of a Sporadic Caudal Regression syndrome with Cleft Lip and Palate RathikaD.Shenoy1 ,SwathiSunilRao 2 ,VijayaD.Shenoy 3 ,VikramShetty 4 1Professor, 2 Assistant Professor,3 Professor & Head, Department of Paediatrics, K.S. Hegde Medical Academy, Nitte University, Deralakatte, Mangalore,4 Director, Department of Paediatrics, Nitte Meenakshi Institute of Craniofacial Surgery, K S Hegde Medical Academy,Nitte University,Deralakatte,Mangalore - 575 018, Karnataka, India. *Corresponding Author : Swathi Sunil Rao , #3/77/1A1, Kanasu, Someshwara, Mangalore-575022, Karnataka, India. Mobile : +91 90085 66112, E-mail : [email protected]. Received : 24-02-2016 Abstract : Review Completed : 01-08-2016 Caudal regression syndrome is a rare congenital malformation and can be associated with lower Accepted : 05-08-2016 limb and renal anomalies. However caudal regression syndrome and cleft lip and palate (CLP) rarely occur together, the prevalence of sacral anomalies in children with CLP being 0.1%. We Keywords : Sacral agenesis; hereby report an eighteen month old male child with sporadic lumbosacral agenesis type III tethered cord; unilateral renal with tethered cord, hypospadias, unilateral renal agenesis and CLP for its rarity. agenesis Access this article online Quick Response Code Introduction Case History Caudal regression syndrome (CRS) (OMIM 600145) also A male child was first seen by us at 18 months with orofacial referred to as caudaldysgenesis is a rare congenital defect, lower limb deformity, urinary and faecal malformation with an incidence of 1 to 2.5 per 100,000 incontinence since birth. He was the only child of healthy, pregnancies.1 It is characterized by variable absence of young, non-consanguineous parents. -
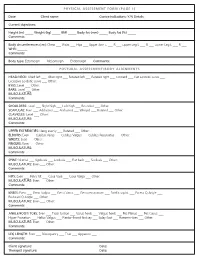
410 PHYSICAL ASSESSMENT FORM (PAGE 1) Date: Client Name
// g 410 PHYSICAL ASSESSMENT FORM (PAGE 1) Date: Client name: Contra-indications: Y/N Details: Current objectives: Height (m) _____ Weight (kg) _____ BMI _____ Body fat (mm) _____ Body fat (%) _____ Comments: APPENDIX 4 SAMPLE FORMS Body circumferences (cm): Chest ___ Waist ___ Hips ___ Upper Arm L ___ R ___ Upper Leg L ___ R ___ Lower Leg L ___ R ___ WHR: ______ Comments: Body type: Ectomorph ___ Mesomorph ___ Endomorph ___ Comments: POSTURAL ASSESSMENT/BODY ALIGNMENTS HEAD/NECK: Tilted left ___ Tilted right ___ Rotated left ___ Rotated right ___ Forward ___ Flat Lordotic curve ___ Excessive Lordotic curve ___ Other: EYES: Level ___ Other: EARS: Level ___ Other: MUSCULATURE: Comments: SHOULDERS: Level ___ Right high ___ Left high ___ Rounded ___ Other: SCAPULAE: Even ___ Adducted ___ Abducted ___ Winged ___ Rotated ___ Other: CLAVICLES: Level ___ Other: MUSCULATURE: Comments: UPPER EXTREMITIES: Hang evenly ___ Rotated ___ Other: ELBOWS: Even ___ Cubitus Varus ___ Cubitus Valgus ___ Cubitus Recurvatus ___ Other: WRISTS: Even ___ Other: FINGERS: Even ___ Other: MUSCULATURE: Comments: SPINE: Normal ___ Kyphosis ___ Lordosis ___ Flat back ___ Scoliosis ___ Other: MUSCULATURE: Even ___ Other: Comments: HIPS: Even ___ Pelvic tilt ___ Coxa Vara ___ Coxa Valga ___ Other: MUSCULATURE: Even ___ Other: Comments: KNEES: Even ___ Genu Valgus ___ Genu Varus ___ Genu recurvatum ___ Patella squint ___ Excess Q Angle ___ Reduced Q Angle ___ Other: MUSCULATURE: Even ___ Other: Comments: ANKLE/FOOT/TOES: Even ___ Tibial torsion ___ Varus heels ___ Valgus -

Differential Diagnosis of Complex Conditions in Paleopathology: a Mutational Spectrum Approach by Elizabeth Lukashal a Thesis
Differential Diagnosis of Complex Conditions in Paleopathology: A Mutational Spectrum Approach by Elizabeth Lukashal A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree of Master of Arts in Public Issues Anthropology Waterloo, Ontario, Canada, 2021 © Elizabeth Lukashal 2021 Author’s Declaration I hereby declare that I am the sole author of this thesis. This is a true copy of the thesis, including any required final revisions, as accepted by my examiners. I understand that my thesis may be made electronically available to the public. ii Abstract The expression of mutations causing complex conditions varies considerably on a scale of mild to severe referred to as a mutational spectrum. Capturing a complete picture of this scale in the archaeological record through the study of human remains is limited due to a number of factors complicating the diagnosis of complex conditions. An array of potential etiologies for particular conditions, and crossover of various symptoms add an extra layer of complexity preventing paleopathologists from confidently attempting a differential diagnosis. This study attempts to address these challenges in a number of ways: 1) by providing an overview of congenital and developmental anomalies important in the identification of mild expressions related to mutations causing complex conditions; 2) by outlining diagnostic features of select anomalies used as screening tools for complex conditions in the medical field ; 3) by assessing how mild/carrier expressions of mutations and conditions with minimal skeletal impact are accounted for and used within paleopathology; and 4) by considering the potential of these mild expressions in illuminating additional diagnostic and environmental information regarding past populations.