L-Carnitine and Acylcarnitines: Mitochondrial Biomarkers for Precision Medicine
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

L-Carnitine, Mecobalamin and Folic Acid Tablets) TRINERVE-LC
For the use of a Registered Medical Practitioner or a Hospital or a Laboratory only (L-Carnitine, Mecobalamin and Folic acid Tablets) TRINERVE-LC 1. Name of the medicinal product Trinerve-LC Tablets 2. Qualitative and quantitative composition Each film- coated tablets contains L-Carnitine…………………….500 mg Mecobalamin……………….1500 mcg Folic acid IP…………………..1.5mg 3. Pharmaceutical form Film- coated tablets 4. Clinical particulars 4.1 Therapeutic indications Vitamin and micronutrient supplementation in the management of chronic disease. 4.2 Posology and method of administration For oral administration only. One tablet daily or as directed by physician. 4.3 Contraindications Hypersensitivity to any constituent of the product. 4.4 Special warnings and precautions for use L-Carnitine The safety and efficacy of oral L-Carnitine has not been evaluated in patients with renal insufficiency. Chronic administration of high doses of oral L-Carnitine in patients with severely compromised renal function or in ESRD patients on dialysis may result in accumulation of the potentially toxic metabolites, trimethylamine (TMA) and trimethylamine-N-oxide (TMAO), since these metabolites are normally excreted in the urine. Mecobalamin Should be given with caution in patients suffering from folate deficiency. The following warnings and precautions suggested with parent form – vitamin B12 The treatment of vitamin B12 deficiency can unmask the symptoms of polycythemia vera. Megaloblastic anemia is sometimes corrected by treatment with vitamin B12. But this can have very serious side effects. Don’t attempt vitamin B12 therapy without close supervision by healthcare provider. Do not take vitamin B12 if Leber’s disease, a hereditary eye disease. -

Carnitine: What Is It? Carnitine
Home Health Information News & Events Research & Funding About ODS Dietary Supplement Fact Sheet: TABLE OF CONTENTS Carnitine: What is it? Carnitine What are recommended intakes Health Professional Other Resources for carnitine? What foods provide carnitine? Carnitine: What is it? Carnitine, derived from an amino acid, is found in nearly all cells of the body. Its name is derived from the Latin Absorption and metabolism of carnus or flesh, as the compound was isolated from meat. Carnitine is the generic term for a number of carnitine compounds that include L-carnitine, acetyl-L-carnitine, and propionyl-L-carnitine [1-2]. When can a carnitine deficiency occur? Carnitine plays a critical role in energy production. It transports long-chain fatty acids into the mitochondria so What are some current issues they can be oxidized ("burned") to produce energy. It also transports the toxic compounds generated out of this and controversies about cellular organelle to prevent their accumulation. Given these key functions, carnitine is concentrated in tissues like carnitine? skeletal and cardiac muscle that utilize fatty acids as a dietary fuel [1-2]. Are there health risks from too much carnitine? The body makes sufficient carnitine to meet the needs of most people. For genetic or medical reasons, some individuals (such as preterm infants), cannot make enough, so for them carnitine is a conditionally essential Carnitine and medication nutrient [1]. interactions Supplemental sources of carnitine What are recommended intakes for carnitine? Carnitine intakes and healthful Healthy children and adults do not need to consume carnitine from food or supplements, as the liver and kidneys diets produce sufficient amounts from the amino acids lysine and methionine to meet daily needs [1-3]. -

Amino Acids Amino Acids
Amino Acids Amino Acids What Are Amino Acids? Essential Amino Acids Non Essential Amino Acids Amino acids are the building blocks of proteins; proteins are made of amino acids. Isoleucine Arginine (conditional) When you ingest a protein your body breaks it down into the individual aminos, Leucine Glutamine (conditional) reorders them, re-folds them, and turns them into whatever is needed by the body at Lysine Tyrosine (conditional) that time. From only 20 amino acids, the body is able to make thousands of unique proteins with different functions. Methionine Cysteine (conditional) Phenylalanine Glycine (conditional) Threonine Proline (conditional) Did You Know? Tryptophan Serine (conditional) Valine Ornithine (conditional) There are 20 different types of amino acids that can be combined to make a protein. Each protein consists of 50 to 2,000 amino acids that are connected together in a specific Histidine* Alanine sequence. The sequence of the amino acids determines each protein’s unique structure Asparagine and its specific function in the body. Asparate Popular Amino Acid Supplements How Do They Benefit Our Health? Acetyl L- Carnitine: As part of its role in supporting L-Lysine: L-Lysine, an essential amino acid, is mental function, Acetyl L-Carnitine may help needed to support proper growth and bone Proteins (amino acids) are needed by your body to maintain muscles, bones, blood, as support memory, attention span and mental development. It can also support immune function. well as create enzymes, neurotransmitters and antibodies, as well as transport and performance. store molecules. N-Acetyl Cysteine: N-Acetyl Cysteine (NAC) is a L-Arginine: L-Arginine is a nonessential amino acid form of the amino acid cysteine. -

Treatment Options for Motor and Non-Motor Symptoms of Parkinson’S Disease
biomolecules Review Treatment Options for Motor and Non-Motor Symptoms of Parkinson’s Disease Frank C. Church Department of Pathology and Laboratory Medicine, The University of North Carolina School of Medicine, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA; [email protected] Abstract: Parkinson’s disease (PD) usually presents in older adults and typically has both motor and non-motor dysfunctions. PD is a progressive neurodegenerative disorder resulting from dopamin- ergic neuronal cell loss in the mid-brain substantia nigra pars compacta region. Outlined here is an integrative medicine and health strategy that highlights five treatment options for people with Parkinson’s (PwP): rehabilitate, therapy, restorative, maintenance, and surgery. Rehabilitating begins following the diagnosis and throughout any additional treatment processes, especially vis-à-vis consulting with physical, occupational, and/or speech pathology therapist(s). Therapy uses daily administration of either the dopamine precursor levodopa (with carbidopa) or a dopamine ago- nist, compounds that preserve residual dopamine, and other specific motor/non-motor-related compounds. Restorative uses strenuous aerobic exercise programs that can be neuroprotective. Maintenance uses complementary and alternative medicine substances that potentially support and protect the brain microenvironment. Finally, surgery, including deep brain stimulation, is pursued when PwP fail to respond positively to other treatment options. There is currently no cure for PD. In conclusion, the best strategy for treating PD is to hope to slow disorder progression and strive to achieve stability with neuroprotection. The ultimate goal of any management program is to improve the quality-of-life for a person with Parkinson’s disease. -

Choline, Inositol, L-Carnitine, Taurine
Ingredients in infant milks Choline, Inositol, L-Carnitine, Taurine Choline Choline is found in a number of forms in human milk, and the choline content of human milk is influenced by maternal choline intake. It is synthesised in the body, and the extent to which it is a required dietary component under normal circumstances is unclear. Choline serves as the precursor for the synthesis of phosphatidyl choline (PC), the main phospholipid in brain, liver and other tissues. PC plays a role in normal membrane composition and signalling processes, lipid metabolism, and normal brain development. As it is essential, and the potential for impaired synthesis or increased needs are not clear, it remains mandatory in infant formula. Following EFSA (2014) recommendations, current regulations specify a minimum choline content of 25mg/100kcal in infant formula, however it is not considered a necessary ingredient in follow-on formula. Inositol Inositol is an essential growth factor which is synthesised in the body but may need to be provided in the diet under certain conditions. Inositol is present in high concentration in human milk and decreases over the course of lactation. Inositol levels in blood are high among neonates, leading to the suggestion that inositol plays an important role in early development (Scientific Committee on Food, 2003) and as the amount of endogenous synthesis in infants is unknown, legislation has previously given minimum and maximum levels for inositol in infant formula suitable from birth. Current regulations reflect the EFSA opinion (2014) that inositol should be added to infant formula at 4mg/100kcal, but that it is not a necessary component of follow-on formula. -

Dietary Supplements Compendium Volume 1
2015 Dietary Supplements Compendium DSC Volume 1 General Notices and Requirements USP–NF General Chapters USP–NF Dietary Supplement Monographs USP–NF Excipient Monographs FCC General Provisions FCC Monographs FCC Identity Standards FCC Appendices Reagents, Indicators, and Solutions Reference Tables DSC217M_DSCVol1_Title_2015-01_V3.indd 1 2/2/15 12:18 PM 2 Notice and Warning Concerning U.S. Patent or Trademark Rights The inclusion in the USP Dietary Supplements Compendium of a monograph on any dietary supplement in respect to which patent or trademark rights may exist shall not be deemed, and is not intended as, a grant of, or authority to exercise, any right or privilege protected by such patent or trademark. All such rights and privileges are vested in the patent or trademark owner, and no other person may exercise the same without express permission, authority, or license secured from such patent or trademark owner. Concerning Use of the USP Dietary Supplements Compendium Attention is called to the fact that USP Dietary Supplements Compendium text is fully copyrighted. Authors and others wishing to use portions of the text should request permission to do so from the Legal Department of the United States Pharmacopeial Convention. Copyright © 2015 The United States Pharmacopeial Convention ISBN: 978-1-936424-41-2 12601 Twinbrook Parkway, Rockville, MD 20852 All rights reserved. DSC Contents iii Contents USP Dietary Supplements Compendium Volume 1 Volume 2 Members . v. Preface . v Mission and Preface . 1 Dietary Supplements Admission Evaluations . 1. General Notices and Requirements . 9 USP Dietary Supplement Verification Program . .205 USP–NF General Chapters . 25 Dietary Supplements Regulatory USP–NF Dietary Supplement Monographs . -

The Relationship Between Choline Bioavailability from Diet, Intestinal Microbiota Composition, and Its Modulation of Human Diseases
nutrients Review The Relationship between Choline Bioavailability from Diet, Intestinal Microbiota Composition, and Its Modulation of Human Diseases Natalia Arias 1,2,*, Silvia Arboleya 3 , Joseph Allison 2, Aleksandra Kaliszewska 2, Sara G. Higarza 1,4 , Miguel Gueimonde 3 and Jorge L. Arias 1,4 1 Instituto de Neurociencias del Principado de Asturias (INEUROPA), 33003 Oviedo, Asturias, Spain; [email protected] (S.G.H.); [email protected] (J.L.A.) 2 Department of Basic and Clinical Neuroscience, Institute of Psychiatry, Psychology and Neuroscience, King’s College London, Denmark Hill, London SE5 8AF, UK; [email protected] (J.A.); [email protected] (A.K.) 3 Department of Microbiology and Biochemistry of Dairy Products, Instituto de Productos Lácteos de Asturias (IPLA-CSIC), 33003 Oviedo, Asturias, Spain; [email protected] (S.A.); [email protected] (M.G.) 4 Laboratory of Neuroscience, Department of Psychology, University of Oviedo, Plaza Feijóo, s/n, 33003 Oviedo, Asturias, Spain * Correspondence: [email protected] Received: 14 July 2020; Accepted: 30 July 2020; Published: 5 August 2020 Abstract: Choline is a water-soluble nutrient essential for human life. Gut microbial metabolism of choline results in the production of trimethylamine (TMA), which, upon absorption by the host is converted into trimethylamine-N-oxide (TMAO) in the liver. A high accumulation of both components is related to cardiovascular disease, inflammatory bowel disease, non-alcoholic fatty liver disease, and chronic kidney disease. However, the relationship between the microbiota production of these components and its impact on these diseases still remains unknown. In this review, we will address which microbes contribute to TMA production in the human gut, the extent to which host factors (e.g., the genotype) and diet affect TMA production, and the colonization of these microbes and the reversal of dysbiosis as a therapy for these diseases. -

Potential Role of L-Carnitine in Autism Spectrum Disorder
Journal of Clinical Medicine Review Potential Role of L-Carnitine in Autism Spectrum Disorder Alina K˛epka 1,† , Agnieszka Ochoci ´nska 1,*,† , Sylwia Chojnowska 2 , Małgorzata Borzym-Kluczyk 3, Ewa Skorupa 1, Małgorzata Kna´s 2 and Napoleon Waszkiewicz 4 1 Department of Biochemistry, Radioimmunology and Experimental Medicine, The Children’s Memorial Health Institute, 04-730 Warsaw, Poland; [email protected] (A.K.); [email protected] (E.S.) 2 Faculty of Health Sciences, Lomza State University of Applied Sciences, 18-400 Lomza, Poland; [email protected] (S.C.); [email protected] (M.K.) 3 Department of Pharmaceutical Biochemistry, Medical University of Bialystok, 15-089 Bialystok, Poland; [email protected] 4 Department of Psychiatry, Medical University of Bialystok, 15-089 Bialystok, Poland; [email protected] * Correspondence: [email protected]; Tel.: +48-22-815-73-01 † These authors are sharing the first place. Both contributed equally to this work. Abstract: L-carnitine plays an important role in the functioning of the central nervous system, and especially in the mitochondrial metabolism of fatty acids. Altered carnitine metabolism, abnormal fatty acid metabolism in patients with autism spectrum disorder (ASD) has been documented. ASD is a complex heterogeneous neurodevelopmental condition that is usually diagnosed in early child- hood. Patients with ASD require careful classification as this heterogeneous clinical category may include patients with an intellectual disability or high functioning, epilepsy, language impairments, or associated Mendelian genetic conditions. L-carnitine participates in the long-chain oxidation of fatty acids in the brain, stimulates acetylcholine synthesis (donor of the acyl groups), stimulates ex- pression of growth-associated protein-43, prevents cell apoptosis and neuron damage and stimulates Citation: K˛epka,A.; Ochoci´nska,A.; neurotransmission. -
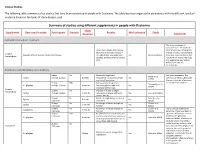
Summary of Studies Using Supplements in Duchenne
Clinical Studies The following table summarizes key studies that have been conducted in people with Duchenne. The table has been organized in accordance with the different levels of evidence based on the types of study designs used. Summary of studies using different supplements in people with Duchenne Study Supplement Dose used in study Participants Steroids Results Well tolerated Study Duration Comments Systematic Review (Level 1 Evidence) This study combined all outcomes from the individual Across the 5 studies listed below, trials listed below. Although the there was an average increase in average increase is encouraging, Creatine Characteristics of included studies listed below strength (8.5%), lean body mass Yes Kley et al. (2013) there was some variation in the monohydrate (0.63kg), and bone mineral density results which means individuals (3%) may experience very modest effects from creatine monohydrate. Randomised controlled trials (Level 2 Evidence) 8 boys No Modest but significant See above comments. The Walter et al. 5 g/day Average 10 years 8 weeks improvement in muscle strength Yes summary of these studies gives (2000) and daily-life activities the best indication of evidence 31 boys 50% Decrease bone breakdown, for creatine monohydrate. Tarnopolsky et al. 0.1 g/kg/day Average 10 years 4 months increased grip strength, and Yes (2004) increased fat free mass Creatine 12 boys No Increase in muscle strength, monohydrate 3 g/day Average 11 years 3 months resistance to fatigue, and bone Yes Louis et al. (2003) mineral density 33 boys No Muscle strength was preserved in Banerjee et al. 5 g/day 8 weeks Yes 3-12 years short term (2010) 50 boys No No change in muscle strength or Escolar et al. -

WO 2015/094925 Al 25 June 2015 (25.06.2015) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2015/094925 Al 25 June 2015 (25.06.2015) P O P C T (51) International Patent Classification: BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, A61K 9/48 (2006.01) DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, (21) International Application Number: KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, PCT/US20 14/06983 1 MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (22) International Filing Date: PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, 11 December 2014 ( 11.12.2014) SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every (26) Publication Language: English kind of regional protection available): ARIPO (BW, GH, (30) Priority Data: GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, 61/917,692 18 December 201 3 (18. 12.2013) US TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, (71) Applicant: CAPSUGEL BELGIUM NV [BE/BE]; Rijk- DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, weg 11, B-2880 Bornem (BE). -

The Efficacy of Antioxidants in Sperm Parameters And
Received: 30 September 2019 | Revised: 27 November 2019 | Accepted: 13 December 2019 DOI: 10.1111/and.13514 INVITED REVIEW The efficacy of antioxidants in sperm parameters and production of reactive oxygen species levels during the freeze- thaw process: A systematic review and meta-analysis Rezvan Bahmyari1 | Morteza Zare2 | Rakesh Sharma3 | Ashok Agarwal3 | Iman Halvaei4 1Department of Midwifery, Shiraz University of Medical Sciences, Shiraz, Iran Abstract 2Nutrition Research Center, School of To investigate the impact of antioxidants in sperm parameters and reduction in reac- Nutrition and Food Sciences, Shiraz tive oxygen species production during the freeze-thaw process. PubMed, Scopus, University of Medical Sciences, Shiraz, Iran 3American Center for Reproductive Web of Science, Embase and Cochrane central library were systematically searched. Medicine, Cleveland Clinic, Cleveland, OH, Of the 1583 articles, 23 studies were selected for data extraction. Our results show USA that antioxidants improved sperm progressive motility (standardised mean difference 4Department of Anatomical Sciences, Faculty of Medical Sciences, Tarbiat (SMD) = 1; 95% CI: 0.62, 1.38; p < .001) and viability (SMD = 1.20; 95% CI: 0.50, Modares University, Tehran, Iran 1.91; p = .001) and reduced sperm DNA fragmentation (SDF) and hydrogen peroxide Correspondence (H2O2) production, but there was no significant improvement in total sperm motility Iman Halvaei, Department of Anatomical after thawing. Acetyl-l-carnitine/l-carnitine, melatonin and catalase had a significant Sciences, Faculty of Medical Sciences, Tarbiat Modares University, Jalal-Ale-Ahmad positive impact on progressive motility. The role of tempol and melatonin in improv- Ave., Nasr Bridge, 14155-331 Tehran, Iran. ing viability was significant compared to other antioxidants. -

Transfer and Metabolism of Carnitine and Carnitine Esters in the in Vitro Perfused Human Placenta
700 SCHMIDT-SOMMERFELD ET AL. and prostaglandin metabolism in zinc deficient pregnant rats. Life Sci istry in Medicine and Biochemistry, Vol 2. Walter de Gmyter, NY, pp 32:247 1-2478 219-226 28. Everett G, Apger J 1983 Effect of zinc deficiency on prostaglandin F2, and E 29. Meydani SN, Dupont J 1982 Effect of zinc deficiency on prostaglandin in the rat. In: Bratter P, Schramel P (eds). Trace Elements-Analytic Chem- synthesis in different organs of rat. J Nutr 6:1098-1104 003 1-3998/85/1907-0700$02.00/0 PEDIATRIC RESEARCH Vol. 19, No. 7, 1985 Copyright O 1985 International Pediatric Research Foundation, Inc. Printed in (I.S. A. Transfer and Metabolism of Carnitine and Carnitine Esters in the in Vitro Perfused Human Placenta EBERHARD SCHMIDT-SOMMERFELD, DUNA PEW, RAVI J. SODHA, MARIETTE PR~GLER, MILAN NOVAK, AND HENNING SCHNEIDER Department of Pediatrics, University of Giessen, Federal Republic of Germany; Division of Perinatal Physiology, Department of Obstetrics, University of Zurich, Switzerland; and Department of Pediatrics, University of Miami, Florida ABSTRAa. The transfer and metabolism of L-carnitine, tremely slow carnitine placental transport has been found in the L-acetylcarnitine, and L-palmitoylcarnitine were studied in sheep in vivo, rapid transfer occurs across the guinea pig placenta the human placenta at term by means of in vitro dual (9). Until now, only indirect evidence exists for carnitine placen- perfusion of a placental lobe. L-Carnitine transfer was 20% tal transfer in the human. A strong correlation has been found that of the freely diffusing antipyrine and 40% that of L- between maternal and umbilical carnitine blood concentrations lysine.