Mouse Asz1 Conditional Knockout Project (CRISPR/Cas9)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Functional Analysis of Structural Variation in the 2D and 3D Human Genome
FUNCTIONAL ANALYSIS OF STRUCTURAL VARIATION IN THE 2D AND 3D HUMAN GENOME by Conor Mitchell Liam Nodzak A dissertation submitted to the faculty of The University of North Carolina at Charlotte in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Bioinformatics and Computational Biology Charlotte 2019 Approved by: Dr. Xinghua Mindy Shi Dr. Rebekah Rogers Dr. Jun-tao Guo Dr. Adam Reitzel ii c 2019 Conor Mitchell Liam Nodzak ALL RIGHTS RESERVED iii ABSTRACT CONOR MITCHELL LIAM NODZAK. Functional analysis of structural variation in the 2D and 3D human genome. (Under the direction of DR. XINGHUA MINDY SHI) The human genome consists of over 3 billion nucleotides that have an average distance of 3.4 Angstroms between each base, which equates to over two meters of DNA contained within the 125 µm3 volume diploid cell nuclei. The dense compaction of chromatin by the supercoiling of DNA forms distinct architectural modules called topologically associated domains (TADs), which keep protein-coding genes, noncoding RNAs and epigenetic regulatory elements in close nuclear space. It has recently been shown that these conserved chromatin structures may contribute to tissue-specific gene expression through the encapsulation of genes and cis-regulatory elements, and mutations that affect TADs can lead to developmental disorders and some forms of cancer. At the population-level, genomic structural variation contributes more to cumulative genetic difference than any other class of mutation, yet much remains to be studied as to how structural variation affects TADs. Here, we study the func- tional effects of structural variants (SVs) through the analysis of chromatin topology and gene activity for three trio families sampled from genetically diverse popula- tions from the Human Genome Structural Variation Consortium. -

Functional Annotation of Genes Overlapping Copy Number Variants in Autistic Patients: Focus on Axon Pathfinding
136 Current Genomics, 2010, 11, 136-145 Functional Annotation of Genes Overlapping Copy Number Variants in Autistic Patients: Focus on Axon Pathfinding Silvia Sbacchi1, Francesco Acquadro2, Ignazio Calò1, Francesco Calì3 and Valentino Romano*,1,3 1Dipartimento di Oncologia Sperimentale e Applicazioni Cliniche, Università degli Studi di Palermo, Palermo; 2Molecular Cytogenetics Group, Centro Nacional de Investigaciones Oncologicas (C.N.I.O.), and Centro de Investiga- ciones de Enfermidades Raras (CIBERER), Madrid, Spain; 3Associazione Oasi Maria SS. (I.R.C.C.S.), Troina (EN), Italy Abstract: We have used Gene Ontology (GO) and pathway analyses to uncover the common functions associated to the genes overlapping Copy Number Variants (CNVs) in autistic patients. Our source of data were four published studies [1- 4]. We first applied a two-step enrichment strategy for autism-specific genes. We fished out from the four mentioned stud- ies a list of 2928 genes overall overlapping 328 CNVs in patients and we first selected a sub-group of 2044 genes after excluding those ones that are also involved in CNVs reported in the Database of Genomic Variants (enrichment step 1). We then selected from the step 1-enriched list a sub-group of 514 genes each of which was found to be deleted or dupli- cated in at least two patients (enrichment step 2). The number of statistically significant processes and pathways identified by the Database for Annotation, Visualization and Integrated Discovery and Ingenuity Pathways Analysis softwares with the step 2-enriched list was significantly higher compared to the step 1-enriched list. In addition, statistically significant GO terms, biofunctions and pathways related to nervous system development and function were exclusively identified by the step 2-enriched list of genes. -

The Identification of 64 Novel Genetic Loci Provides an Expanded View on the Genetic Architecture of Coronary Artery Disease
University of Groningen Identification of 64 Novel Genetic Loci Provides an Expanded View on the Genetic Architecture of Coronary Artery Disease van der Harst, Pim; Verweij, Niek Published in: Circulation research DOI: 10.1161/CIRCRESAHA.117.312086 IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite from it. Please check the document version below. Document Version Publisher's PDF, also known as Version of record Publication date: 2018 Link to publication in University of Groningen/UMCG research database Citation for published version (APA): van der Harst, P., & Verweij, N. (2018). Identification of 64 Novel Genetic Loci Provides an Expanded View on the Genetic Architecture of Coronary Artery Disease. Circulation research, 122(3), 433-443. https://doi.org/10.1161/CIRCRESAHA.117.312086 Copyright Other than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons). The publication may also be distributed here under the terms of Article 25fa of the Dutch Copyright Act, indicated by the “Taverne” license. More information can be found on the University of Groningen website: https://www.rug.nl/library/open-access/self-archiving-pure/taverne- amendment. Take-down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. -

Homology-Based Negative Data Sampling Method for Genome-Scale Reconstruction of Human Protein–Protein Interaction Networks
International Journal of Molecular Sciences Article Neglog: Homology-Based Negative Data Sampling Method for Genome-Scale Reconstruction of Human Protein–Protein Interaction Networks Suyu Mei 1,* and Kun Zhang 2,* 1 Software College, Shenyang Normal University, Shenyang 110034, China 2 Bioinformatics Facility of Xavier NIH RCMI Cancer Research Center, Department of Computer Science, Xavier University of Louisiana, New Orleans, LA 70125, USA * Correspondence: [email protected] (S.M.); [email protected] (K.Z.) Received: 27 September 2019; Accepted: 11 October 2019; Published: 12 October 2019 Abstract: Rapid reconstruction of genome-scale protein–protein interaction (PPI) networks is instrumental in understanding the cellular processes and disease pathogenesis and drug reactions. However, lack of experimentally verified negative data (i.e., pairs of proteins that do not interact) is still a major issue that needs to be properly addressed in computational modeling. In this study, we take advantage of the very limited experimentally verified negative data from Negatome to infer more negative data for computational modeling. We assume that the paralogs or orthologs of two non-interacting proteins also do not interact with high probability. We coin an assumption as “Neglog” this assumption is to some extent supported by paralogous/orthologous structure conservation. To reduce the risk of bias toward the negative data from Negatome, we combine Neglog with less biased random sampling according to a certain ratio to construct training data. L2-regularized logistic regression is used as the base classifier to counteract noise and train on a large dataset. Computational results show that the proposed Neglog method outperforms pure random sampling method with sound biological interpretability. -

Specification and Epigenetic Resetting of the Pig Germline Exhibit Conservation with the Human Lineage
bioRxiv preprint doi: https://doi.org/10.1101/2020.08.07.241075; this version posted August 7, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Specification and epigenetic resetting of the pig germline exhibit conservation with the human lineage Qifan Zhu1#, Fei Sang2#, Sarah Withey1^§, Walfred Tang3,4^, Sabine Dietmann3^, Doris Klisch1, Priscila Ramos-Ibeas1§, Haixin Zhang1§, Cristina E. Requena5,6, Petra Hajkova5,6, Matt Loose2, M. Azim Surani3,4,7*, Ramiro Alberio1* 1 School of Biosciences, University of Nottingham, Sutton Bonington Campus, LE12 5RD, UK. 2 School of Life Sciences, University of Nottingham, Nottingham, NG7 2RD, UK. 3 Wellcome Trust/Cancer Research UK Gurdon Institute, University of Cambridge, Tennis Court Road, Cambridge CB2 1QN, UK. 4 Department of Physiology, Development and Neuroscience, University of Cambridge, Downing Street, Cambridge CB2 3DY, UK. 5 MRC London Institute of Medical Sciences (LMS), London, UK. 6 Institute of Clinical Sciences (ICS), Faculty of Medicine, Imperial College London, London, UK. 7 Wellcome Trust Medical Research Council Stem Cell Institute, University of Cambridge, Tennis Court Road, Cambridge CB2 1QR, UK § Current address: P.R-I.: Animal Reproduction Department, National Institute for Agricultural and Food Research and Technology, Madrid 28040, Spain; S.W.: Stem Cell Engineering Group, Australian Institute for Bioengineering and Nanotechnology, University of Queensland, Building 75, St Lucia, QLD 4072, Australia. # Equal contribution, ^ Equal contribution * Co-corresponding authors Email addresses of corresponding authors: [email protected]; [email protected] Lead contact: Ramiro Alberio 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.08.07.241075; this version posted August 7, 2020. -

Chromatin Dynamics and the Role of G9a in Gene Regulation And
RESEARCH ARTICLE Chromatin dynamics and the role of G9a in gene regulation and enhancer silencing during early mouse development Jan J Zylicz1,2,3, Sabine Dietmann3, Ufuk Gu¨ nesdogan1,2, Jamie A Hackett1,2, Delphine Cougot1,2, Caroline Lee1,2, M Azim Surani1,2* 1Wellcome Trust/Cancer Research United Kingdom Gurdon Institute, University of Cambridge, Cambridge, United Kingdom; 2Department of Physiology, Development and Neuroscience, University of Cambridge, Cambridge, United Kingdom; 3Wellcome Trust/Medical Research Council Stem Cell Institute, University of Cambridge, Cambridge, United Kingdom Abstract Early mouse development is accompanied by dynamic changes in chromatin modifications, including G9a-mediated histone H3 lysine 9 dimethylation (H3K9me2), which is essential for embryonic development. Here we show that genome-wide accumulation of H3K9me2 is crucial for postimplantation development, and coincides with redistribution of enhancer of zeste homolog 2 (EZH2)-dependent histone H3 lysine 27 trimethylation (H3K27me3). Loss of G9a or EZH2 results in upregulation of distinct gene sets involved in cell cycle regulation, germline development and embryogenesis. Notably, the H3K9me2 modification extends to active enhancer elements where it promotes developmentally-linked gene silencing and directly marks promoters and gene bodies. This epigenetic mechanism is important for priming gene regulatory networks for critical cell fate decisions in rapidly proliferating postimplantation epiblast cells. DOI: 10.7554/eLife.09571.001 *For correspondence: a.surani@ gurdon.cam.ac.uk Competing interests: The Introduction authors declare that no Early mammalian development progresses through a series of landmark events that are regulated by competing interests exist. transcriptional and epigenetic mechanisms. Accordingly, establishment of the pluripotent inner cell mass (ICM) in E3.5 blastocysts is linked with global DNA hypomethylation (Figure 1A) Funding: See page 22 (Hackett et al., 2013a; Smith et al., 2012; Wang et al., 2014). -
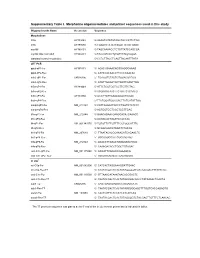
Supplementary Table I. Morpholino Oligonucleotides and Primer Sequences Used in This Study
Supplementary Table I. Morpholino oligonucleotides and primer sequences used in this study Oligonucleotide Name Accession Sequence Morpholinos tlr5a AY389449 5'-AAAGTGTATGTAGCTGCCATTCTGG tlr5b AY389450 5'-TGAATGTATATCCCATTCTGTGAGC myd88 AY388401 5'-TAGCAAAACCTCTGTTATCCAGCGA myd88 5bp mismatch AY388401 5'-TAcCAtAACCTgTGTTATCgAGgGA standard control morpholino 5'-CCTCTTACCTCAGTTACAATTTATA qRT-PCR ppial-qP1-Fw AY391451 5’- ACACTGAAACACGGAGGCAAAG ppial-qP2-Rev 5’- CATCCACAACCTTCCCGAACAC irak3-qP1-Fw CK026195 5’- TGAGGTCTACTGTGGACGATGG irak3-qP2-Rev 5’- ATGTTAGGATGCTGGTTGAGTTGG tlr5a-qP1-Fw AY389449 5’-ATTCTGGTGGTGCTTGTTGTAG tlr5a-qP2-Rev 5’-ACGAGGTAACTTCTGTTCTCAATG tlr5b-qP3-Fw AY389450 5’-GCGTTGTTGAAGAGGCTGGAC tlr5b-qP4-Rev 5’-TTCTGGATGGCCACTTCTCATATTGG mmp9-qP3-Fw NM_213123 5’-CATTAAAGATGCCCTGATGTATCCC mmp9-qP4-Rev 5’-AGTGGTGGTCCGTGGTTGAG il1b-qP1-Fw NM_212844 5’-GAACAGAATGAAGCACATCAAACC il1b-qP2-Rev 5’-ACGGCACTGAATCCACCAC il8-qP1-Fw XM_001342570 5’-TGTGTTATTGTTTTCCTGGCATTTC il8-qP2-Rev 5’-GCGACAGCGTGGATCTACAG ifn1-qP3-Fw NM_207640 5’- TTAATACACGCAAAGATGAGAACTC ifn1-qP4-Rev 5’- GCCAAGCCATTCGCAAGTAG tnfa-qP5-Fw NM_212829 5’- AGACCTTAGACTGGAGAGATGAC tnfa-qP6-Rev 5’- CAAAGACACCTGGCTGTAGAC cxcl-C1c-qP1-Fw NM_001115060 5’- GGCATTCACACCCAAAGCG cxcl-C1c-qP2_Rev 5’- GCGAGCACGATTCACGAGAG * In situ ccl-C5a-Fw NM_001082906 5’- CATCACTAGGAAAGGATTGAAC ccl-C5a-Rev-T7 5’- TAATACGACTCACTATAGGGGATGTCAAAGACTTTATTCAC cxcl-C1c-Fw NM_001115060 5’- GTTAAACATAAATAACACCGACTC cxcl-C1c-Rev-T7 5’- TAATACGACTCACTATAGGGACACCCTATAAAACTGAGTA irak3-Fw CK026195 5’- CAGTGAGAGAGGCATGAAACATC -

VIEW Open Access Restricting Retrotransposons: a Review John L
Goodier Mobile DNA (2016) 7:16 DOI 10.1186/s13100-016-0070-z REVIEW Open Access Restricting retrotransposons: a review John L. Goodier Abstract Retrotransposons have generated about 40 % of the human genome. This review examines the strategies the cell has evolved to coexist with these genomic “parasites”, focussing on the non-long terminal repeat retrotransposons of humans and mice. Some of the restriction factors for retrotransposition, including the APOBECs, MOV10, RNASEL, SAMHD1, TREX1, and ZAP, also limit replication of retroviruses, including HIV, and are part of the intrinsic immune system of the cell. Many of these proteins act in the cytoplasm to degrade retroelement RNA or inhibit its translation. Some factors act in the nucleus and involve DNA repair enzymes or epigenetic processes of DNA methylation and histone modification. RISC and piRNA pathway proteins protect the germline. Retrotransposon control is relaxed in some cell types, such as neurons in the brain, stem cells, and in certain types of disease and cancer, with implications for human health and disease. This review also considers potential pitfalls in interpreting retrotransposon-related data, as well as issues to consider for future research. Keywords: Alu, Autoimmunity, Epigenetics, LINE-1, Methylation, Restriction, Retrovirus, RNAi, SINE, SVA Background highly mutated. However, some intracisternal A-particle Sixty-five years on from Barbara McClintock’s seminal (IAP) and Etn/MusD family LTR elements remain inser- discovery of mobile DNA [1] we now understand that tionally active in mice [8], and formation of infective virions genomes are dynamic and changeable, with transposable by recombination or phenotypic mixing of intact proteins elements (TEs) being major contributors to their fluidity. -

Mrna Expression in Human Leiomyoma and Eker Rats As Measured by Microarray Analysis
Table 3S: mRNA Expression in Human Leiomyoma and Eker Rats as Measured by Microarray Analysis Human_avg Rat_avg_ PENG_ Entrez. Human_ log2_ log2_ RAPAMYCIN Gene.Symbol Gene.ID Gene Description avg_tstat Human_FDR foldChange Rat_avg_tstat Rat_FDR foldChange _DN A1BG 1 alpha-1-B glycoprotein 4.982 9.52E-05 0.68 -0.8346 0.4639 -0.38 A1CF 29974 APOBEC1 complementation factor -0.08024 0.9541 -0.02 0.9141 0.421 0.10 A2BP1 54715 ataxin 2-binding protein 1 2.811 0.01093 0.65 0.07114 0.954 -0.01 A2LD1 87769 AIG2-like domain 1 -0.3033 0.8056 -0.09 -3.365 0.005704 -0.42 A2M 2 alpha-2-macroglobulin -0.8113 0.4691 -0.03 6.02 0 1.75 A4GALT 53947 alpha 1,4-galactosyltransferase 0.4383 0.7128 0.11 6.304 0 2.30 AACS 65985 acetoacetyl-CoA synthetase 0.3595 0.7664 0.03 3.534 0.00388 0.38 AADAC 13 arylacetamide deacetylase (esterase) 0.569 0.6216 0.16 0.005588 0.9968 0.00 AADAT 51166 aminoadipate aminotransferase -0.9577 0.3876 -0.11 0.8123 0.4752 0.24 AAK1 22848 AP2 associated kinase 1 -1.261 0.2505 -0.25 0.8232 0.4689 0.12 AAMP 14 angio-associated, migratory cell protein 0.873 0.4351 0.07 1.656 0.1476 0.06 AANAT 15 arylalkylamine N-acetyltransferase -0.3998 0.7394 -0.08 0.8486 0.456 0.18 AARS 16 alanyl-tRNA synthetase 5.517 0 0.34 8.616 0 0.69 AARS2 57505 alanyl-tRNA synthetase 2, mitochondrial (putative) 1.701 0.1158 0.35 0.5011 0.6622 0.07 AARSD1 80755 alanyl-tRNA synthetase domain containing 1 4.403 9.52E-05 0.52 1.279 0.2609 0.13 AASDH 132949 aminoadipate-semialdehyde dehydrogenase -0.8921 0.4247 -0.12 -2.564 0.02993 -0.32 AASDHPPT 60496 aminoadipate-semialdehyde -

Transposon Regulation Upon Dynamic Loss of DNA Methylation Marius Walter
Transposon regulation upon dynamic loss of DNA methylation Marius Walter To cite this version: Marius Walter. Transposon regulation upon dynamic loss of DNA methylation. Development Biology. Université Pierre et Marie Curie - Paris VI, 2015. English. NNT : 2015PA066672. tel-01375673 HAL Id: tel-01375673 https://tel.archives-ouvertes.fr/tel-01375673 Submitted on 3 Oct 2016 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Université Pierre et Marie Curie Génétique et Biologie du École doctorale Complexité du Vivant developpement Institut Curie` CNRS U934 - INSERM UMR 3215 Transposon regulation upon dynamic loss of DNA methylation Thèse de Doctorat de Biologie présentée par Marius WALTER et dirigée par Déborah BOURC'HIS Présentée et soutenue le 10 Décembre 2015 devant un jury composé de: Dr. Antonin Morillon President du jury Pr. Wolf Reik Rapporteur Pr. Donal O'Carroll Rapporteur Dr. Vincent Colot Examinateur Dr. Michael Weber Examinateur Dr. Déborah Bourc'his Directrice de thèse 2 “There is large amount of evidence which suggests, but does not prove, -
Intronic Enhancers Coordinate Epithelial-Specific Looping of the Active CFTR Locus
Intronic enhancers coordinate epithelial-specific looping of the active CFTR locus Christopher J. Otta, Neil P. Blackledgea,1,2, Jenny L. Kerschnera,1, Shih-Hsing Leira, Gregory E. Crawfordb, Calvin U. Cottonc, and Ann Harrisa,3 aHuman Molecular Genetics Program, Children’s Memorial Research Center, Northwestern University Feinberg School of Medicine, 2300 Children’s Plaza #211, Chicago, IL 60614; bInstitute for Genome Sciences and Policy, Duke University, Durham, NC 27708; and cDepartments of Pediatrics, Physiology, and Biophysics, Case Western Reserve University School of Medicine, Cleveland, OH 44106 Edited by Bing Ren, University of California–San Diego School of Medicine, La Jolla, CA, and accepted by the Editorial Board September 22, 2009 (received for review January 28, 2009) The regulated expression of large human genes can depend on conformation to facilitate expression. The CFTR gene encom- long-range interactions to establish appropriate three-dimensional passes 189 kb at human chromosome 7q31.2 and mutations structures across the locus. The cystic fibrosis transmembrane within it cause the common genetic disease cystic fibrosis (CF) conductance regulator (CFTR) gene, which encompasses 189 kb of (9). CFTR encodes a membrane-associated chloride ion channel genomic DNA, shows a complex pattern of expression with both that is expressed at the highest levels in chloride-secreting spatial and temporal regulation. The flanking loci, ASZ1 and epithelial cells of the small intestine, pancreas, and male genital CTTNBP2, show very different tissue-specific expression. The mech- duct, and at lower levels in the respiratory epithelium and certain anisms governing control of CFTR expression remain poorly un- other sites (10–14). -
Searching for Rare Variants Associated with Osahs-Related Phenotypes
SEARCHING FOR RARE VARIANTS ASSOCIATED WITH OSAHS-RELATED PHENOTYPES THROUGH PEDIGREES by JINGJING LIANG Dissertation Advisor: Dr. Xiaofeng Zhu Department of Population and Quantitative Health Sciences CASE WESTERN RESERVE UNIVERSITY May 29, 2019 CASE WESTERN RESERVE UNIVERSITY SCHOOL OF GRADUATE STUDIES We hereby approve the thesis/dissertation of Jingjing Liang candidate for the degree of Ph.D Committee Chair Scott M. Williams Committee Member Jonathan L. Haines Committee Member Xiaofeng Zhu Committee Member Rong Xu Committee Member Curtis M. Tatsuoka Date of Defense January 29, 2019 *We also certify that written approval has been obtained for any proprietary material contained therein. 1 Table of Contents CHAPTER 1: LITERATURE REVIEW AND SPECIFIC AIMS ………………14 1.1 Obstructive sleep apnea-hypopnea syndrome …………………………………..14 1.2 AHI and SpO2 …………………………………………………………………...16 1.3 Rare variants and missing heritability …………………………………………..22 1.4 Rare variant association analysis ...………………………………………………24 1.5 Rare variant test using pedigree………………………………………………….27 1.6 Annotating variants in genetic regions ………………………………………….29 1.7 Mendelian randomization………………………………………………………..32 1.8 Specific aims ……………………………………………………………………36 CHAPTER 2: IDENTIFYING LOW FREQUENCY AND RARE VARIANTS ASSOCIATED WITH AVSPO2S USING PEDIGREES .…….………38 2.1 Introduction ……………………………………………………………………...38 2.2 Material and methods……………………………………………………………..42 2.2.1 Description of study samples…..……………………………………………..42 2.2.2 Overview of the method………………………………………………………45 2.2.3 Primary