FDA Briefing Document: Pfizer-Biontech COVID-19 Vaccine
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gavi's Vaccine Investment Strategy
Gavi’s Vaccine Investment Strategy Deepali Patel THIRD WHO CONSULTATION ON GLOBAL ACTION PLAN FOR INFLUENZA VACCINES (GAP III) Geneva, Switzerland, 15-16 November 2016 www.gavi.org Vaccine Investment Strategy (VIS) Evidence-based approach to identifying new vaccine priorities for Gavi support Strategic investment Conducted every 5 years decision-making (rather than first-come- first-serve) Transparent methodology Consultations and Predictability of Gavi independent expert advice programmes for long- term planning by Analytical review of governments, industry evidence and modelling and donors 2 VIS is aligned with Gavi’s strategic cycle and replenishment 2011-2015 Strategic 2016-2020 Strategic 2021-2025 period period 2008 2011 2012 2013 2014 2015 2016 2017 2018 2019 2020 2021 2022 2023 2024 2025 RTS,S pilot funding decision VIS #1 VIS #2 VIS #3 MenA, YF mass campaigns, JE, HPV Cholera stockpile, Mid 2017 : vaccine ‘long list’ Rubella, Rabies/cholera studies, Oct 2017 : methodology Typhoid Malaria – deferred Jun 2018 : vaccine shortlist conjugate Dec 2018 : investment decisions 3 VIS process Develop Collect data Develop in-depth methodology and Apply decision investment decision framework for cases for framework with comparative shortlisted evaluation analysis vaccines criteria Phase I Narrow long list Phase II Recommend for Identify long list to higher priority Gavi Board of vaccines vaccines approval of selected vaccines Stakeholder consultations and independent expert review 4 Evaluation criteria (VIS #2 – 2013) Additional Health Implementation -

Risk Factors, Seroprevalence and Infectivity of Hepatitis B Virus
ISSN: 2474-3658 Ogbonna et al. J Infect Dis Epidemiol 2021, 7:186 DOI: 10.23937/2474-3658/1510186 Volume 7 | Issue 1 Journal of Open Access Infectious Diseases and Epidemiology Research ArticlE Risk Factors, Seroprevalence and Infectivity of Hepatitis B Virus amongst Children Resident in Orphanages in a Developing Country Chioma Paulina Ogbonna, MBBS, FMCPaed1, Christian Chukwukere Ogoke, MBBS, FWACP2* , Anthony Nnaemeka Ikefuna, MBBS, FMCPaed, FRCP3,4, Tochukwu 1 Check for Chukwukadibia Ezeofor, MBBS, FMCpaed, FRCP , Emeka Charles Nwolisa, MBBS, updates FWACP, FRCP5, Franklin Chime Emerenini, MBBch, FMCpaed1, and Christopher Bismarck Eke MBBS, FWACP4,6 1Consultant Paediatrician, Department of Paediatrics, Federal Medical Centre, Owerri, Nigeria 2Paediatric Neurology Unit, Department of Paediatrics, King Fahad Central Hospital, Abu Arish 82666, Kingdom of Saudi Arabia 3Professor of Paediatrics, Faculty of Medical Sciences, Department of Paediatrics, University of Nigeria, Nigeria 4Consultant Paediatrician, University of Nigeria Teaching Hospital, Nigeria 5Chief Consultant Paediatrician, Federal Medical Centre, Owerri, Nigeria 6Senior Lecturer, Department of Paediatrics, University of Nigeria, Nigeria *Corresponding authors: Dr. Christian Chukwukere Ogoke, Paediatric Neurology Unit, Department of Paediatrics, King Fahad Central Hospital, Abu Arish 82666, Kingdom of Saudi Arabia Abstract Results: Seroprevalence of HBsAg in children in orphanag- es was 20.0% versus 10.7% in the controls (P = 0.031). In- Background: Hepatitis B infection (HBV) remains a signifi- fectivity of HBV was higher in subjects (42.9%) than controls cant clinical and public health problem and is hyperendemic (20.0%) (P = 0.245). Sharing of towel (P = 0.014) sharing of in Nigeria. In highly endemic regions, infections spread from barbing devices (P = 0.001) and circumcision (P = 0.005) mother to child, or by horizontal transmission, with the bur- were the significant risk factors for Hepatitis B virus infec- den of infection being highest in under-fives. -

Scientific Committee Animal Health and Animal Welfare
EUROPEAN COMMISSION HEALTH and CONSUMER PROTECTION DIRECTORATE-GENERAL Directorate C - Scientific Opinions C2 - Management of scientific committees; scientific co-operation and networks Diagnostic Techniques and Vaccines for Foot-and-Mouth Disease, Classical Swine Fever, Avian Influenza and some other important OIE List A Diseases Report of the Scientific Committee on Animal Health and Animal Welfare Adopted 24-25th April 2003 TABLE OF CONTENTS 1. ABBREVIATIONS.....................................................................................................6 2. MANDATE.................................................................................................................9 3. BACKGROUND.........................................................................................................9 4. PREAMBLE..............................................................................................................10 5. RECENT DEVELOPMENTS IN THE DIAGNOSIS OF INFECTIOUS DISEASES ................................................................................................................14 5.1. Introduction .....................................................................................................14 5.2. Screening of animal products ..........................................................................19 5.3. Nucleic acid amplification methods ................................................................19 5.3.1. Extraction of nucleic acid from the test sample ................................20 5.3.2. Target amplification -

FDA Oncology Experience in Innovative Adaptive Trial Designs
Innovative Adaptive Trial Designs Rajeshwari Sridhara, Ph.D. Director, Division of Biometrics V Office of Biostatistics, CDER, FDA 9/3/2015 Sridhara - Ovarian cancer workshop 1 Fixed Sample Designs • Patient population, disease assessments, treatment, sample size, hypothesis to be tested, primary outcome measure - all fixed • No change in the design features during the study Adaptive Designs • A study that includes a prospectively planned opportunity for modification of one or more specified aspects of the study design and hypotheses based on analysis of data (interim data) from subjects in the study 9/3/2015 Sridhara - Ovarian cancer workshop 2 Bayesian Designs • In the Bayesian paradigm, the parameter measuring treatment effect is regarded as a random variable • Bayesian inference is based on the posterior distribution (Bayes’ Rule – updated based on observed data) – Outcome adaptive • By definition adaptive design 9/3/2015 Sridhara - Ovarian cancer workshop 3 Adaptive Designs (Frequentist or Bayesian) • Allows for planned design modifications • Modifications based on data accrued in the trial up to the interim time • Unblinded or blinded interim results • Control probability of false positive rate for multiple options • Control operational bias • Assumes independent increments of information 9/3/2015 Sridhara - Ovarian cancer workshop 4 Enrichment Designs – Prognostic or Predictive • Untargeted or All comers design: – post-hoc enrichment, prospective-retrospective designs – Marker evaluation after randomization (example: KRAS in cetuximab -
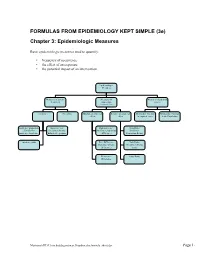
FORMULAS from EPIDEMIOLOGY KEPT SIMPLE (3E) Chapter 3: Epidemiologic Measures
FORMULAS FROM EPIDEMIOLOGY KEPT SIMPLE (3e) Chapter 3: Epidemiologic Measures Basic epidemiologic measures used to quantify: • frequency of occurrence • the effect of an exposure • the potential impact of an intervention. Epidemiologic Measures Measures of disease Measures of Measures of potential frequency association impact (“Measures of Effect”) Incidence Prevalence Absolute measures of Relative measures of Attributable Fraction Attributable Fraction effect effect in exposed cases in the Population Incidence proportion Incidence rate Risk difference Risk Ratio (Cumulative (incidence density, (Incidence proportion (Incidence Incidence, Incidence hazard rate, person- difference) Proportion Ratio) Risk) time rate) Incidence odds Rate Difference Rate Ratio (Incidence density (Incidence density difference) ratio) Prevalence Odds Ratio Difference Macintosh HD:Users:buddygerstman:Dropbox:eks:formula_sheet.doc Page 1 of 7 3.1 Measures of Disease Frequency No. of onsets Incidence Proportion = No. at risk at beginning of follow-up • Also called risk, average risk, and cumulative incidence. • Can be measured in cohorts (closed populations) only. • Requires follow-up of individuals. No. of onsets Incidence Rate = ∑person-time • Also called incidence density and average hazard. • When disease is rare (incidence proportion < 5%), incidence rate ≈ incidence proportion. • In cohorts (closed populations), it is best to sum individual person-time longitudinally. It can also be estimated as Σperson-time ≈ (average population size) × (duration of follow-up). Actuarial adjustments may be needed when the disease outcome is not rare. • In an open populations, Σperson-time ≈ (average population size) × (duration of follow-up). Examples of incidence rates in open populations include: births Crude birth rate (per m) = × m mid-year population size deaths Crude mortality rate (per m) = × m mid-year population size deaths < 1 year of age Infant mortality rate (per m) = × m live births No. -

Whats Good Events Guide December 5-8 2019 Gainesville and Alachua
WHAT’S GOOD. ALACHUA | ARCHER | GAINESVILLE | HAWTHORNE | HIGH SPRINGS | LA CROSSE | MICANOPY | NEWBERRY | WALDO Plan your weekend with the official events guide from Visit Gainesville, Alachua County December 5-8, 2019 Enjoy a Magical Holiday Theatrical Performance for the Entire Family at Spirit of the Horse Friday, December 6 – Saturday, December 7, 7 p.m. – 9 p.m., Sunday, December 8, 6 p.m. – 8 p.m. | Alachua County Agriculture and Equestrian Center 23100 W Newberry Rd., Newberry, FL 32669 Experience an inspiring holiday story that is sure to delight and entertain. This show is in its tenth year, enjoyed by audiences around the country, and 2019 marks the premiere of Spirit of the Horse in Florida. Admission is FREE FOR VETERANS with valid ID. Food available from BubbaQue’s BBQ. Browse Unique Gifts from More Than 200 Vendors at the 51st Annual Craft Festival Saturday, December 7 – Sunday, December 8, 10 a.m. – 5 p.m. | O’Connell Center 250 Gale Lemerand Dr., Gainesville, FL 32611 Find something special for yourself or that hard to shop for person in your life. The festival has been a holiday tradition for the past 51 years and is the largest indoor craft fair in North Central Florida. Watch Santa Arrive by Helicopter at Operation Santa Delivery Saturday, December 7, 10 a.m. – 1 p.m. | Santa Fe College North Field 3700 NW 91st St., Gainesville, FL 32606 Santa will visit Gainesville not by reindeer and sleigh, but by helicopter. Enjoy games, music, food and fun activities. Helicopter Photo by the Gainesville Sun. -

Incidence and Secondary Transmission of SARS-Cov-2 Infections in Schools
Prepublication Release Incidence and Secondary Transmission of SARS-CoV-2 Infections in Schools Kanecia O. Zimmerman, MD; Ibukunoluwa C. Akinboyo, MD; M. Alan Brookhart, PhD; Angelique E. Boutzoukas, MD; Kathleen McGann, MD; Michael J. Smith, MD, MSCE; Gabriela Maradiaga Panayotti, MD; Sarah C. Armstrong, MD; Helen Bristow, MPH; Donna Parker, MPH; Sabrina Zadrozny, PhD; David J. Weber, MD, MPH; Daniel K. Benjamin, Jr., MD, PhD; for The ABC Science Collaborative DOI: 10.1542/peds.2020-048090 Journal: Pediatrics Article Type: Regular Article Citation: Zimmerman KO, Akinboyo IC, Brookhart A, et al. Incidence and secondary transmission of SARS-CoV-2 infections in schools. Pediatrics. 2021; doi: 10.1542/peds.2020- 048090 This is a prepublication version of an article that has undergone peer review and been accepted for publication but is not the final version of record. This paper may be cited using the DOI and date of access. This paper may contain information that has errors in facts, figures, and statements, and will be corrected in the final published version. The journal is providing an early version of this article to expedite access to this information. The American Academy of Pediatrics, the editors, and authors are not responsible for inaccurate information and data described in this version. Downloaded from©2021 www.aappublications.org/news American Academy by of guest Pediatrics on September 27, 2021 Prepublication Release Incidence and Secondary Transmission of SARS-CoV-2 Infections in Schools Kanecia O. Zimmerman, MD1,2,3; Ibukunoluwa C. Akinboyo, MD1,2; M. Alan Brookhart, PhD4; Angelique E. Boutzoukas, MD1,2; Kathleen McGann, MD2; Michael J. -

Package Insert
HIGHLIGHTS OF PRESCRIBING INFORMATION ----------------------------- CONTRAINDICATIONS ------------------------------------- These highlights do not include all the information needed to use ® • History of severe allergic reactions (e.g., anaphylaxis) to egg proteins, or any FLUVIRIN (Influenza Virus Vaccine) safely and effectively. See full ® ® component of FLUVIRIN , or life-threatening reactions to previous influenza prescribing information for FLUVIRIN . vaccinations. (4.1, 11) FLUVIRIN® (Influenza Virus Vaccine) Suspension for Intramuscular Injection ---------------------- WARNINGS AND PRECAUTIONS ------------------------------ 2017-2018 Formula • If Guillain-Barré syndrome has occurred within 6 weeks of receipt of prior Initial US Approval: 1988 influenza vaccine, the decision to give FLUVIRIN® should be based on -------------------------- INDICATIONS AND USAGE ---------------------------------- careful consideration of the potential benefits and risks. (5.1) ® • FLUVIRIN is an inactivated influenza virus vaccine indicated for active • Immunocompromised persons may have a reduced immune response to immunization of persons 4 years of age and older against influenza disease FLUVIRIN®. (5.2) caused by influenza virus subtypes A and type B contained in the vaccine (1). • The tip caps of the FLUVIRIN® prefilled syringes may contain natural rubber • FLUVIRIN® is not indicated for children less than 4 years of age because latex which may cause allergic reactions in latex sensitive individuals. there is evidence of diminished immune response -

Daily Unemployment Compensation Data
DISTRICT OF COLUMBIA DOES DISTRICT OF COLUMBIA DEPARTMENT OF DAILY UNEMPLOYMENT EMPLOYMENT SERVICES COMPENSATION DATA Preliminary numbers as of March 4, 2021.* Telephone Date Online Claims Daily Total Running Total Claims March 13, 2020 310 105 415 415 March 14, 2020 213 213 628 March 15, 2020 410 410 1,038 March 16, 2020 1,599 158 1,757 2,795 March 17, 2020 2,541 219 2,760 5,555 March 18, 2020 2,740 187 2,927 8,482 March 19, 2020 2,586 216 2,802 11,284 March 20, 2020 2,726 205 2,931 14,215 March 21, 2020 1,466 1,466 15,681 March 22, 2020 1,240 1,240 16,921 March 23, 2020 2,516 296 2,812 19,733 March 24, 2020 2,156 236 2.392 22,125 March 25, 2020 2,684 176 2,860 24,985 March 26, 2020 2,842 148 2,990 27,975 March 27, 2020 2,642 157 2,799 30,774 March 28, 2020 1,666 25 1,691 32,465 March 29, 2020 1,547 1,547 34,012 March 30, 2020 2,831 186 3,017 37,029 March, 31, 2020 2,878 186 3,064 40,093 April 1, 2020 2,569 186 2,765 42,858 April 2, 2020 2,499 150 2,649 45,507 April 3, 2020 2,071 300 2,371 47,878 April 4, 2020 1,067 14 1,081 48,959 April 5, 2020 1,020 1,020 49,979 April 6, 2020 2,098 155 2,253 52,232 April 7, 2020 1,642 143 1,715 54,017 April 8, 2020 1,486 142 1,628 55,645 *Recalculated and updated daily DISTRICT OF COLUMBIA Telephone DODISTRICT OF CEOLUMBIASDate Online Claims Daily Total Running Total DEPARTMENT OF DAILY UNEMPLOYMENTClaims EMPLOYMENT SERVICES April 9, 2020 1,604 111 1,715 57,360 April 10, 2020 COMPENSATION1,461 119 1,580 DATA58,940 April 11, 2020 763 14 777 59,717 April 12, 2020 698 698 60,415 April 13, 2020 1,499 104 -

Third Quarter 2020
March 31, 2020 Third Quarter 2020 Corporate update and financial results November 10, 2020 Forward-looking statements Various statements in this slide presentation concerning the future expectations of BioNTech, its plans and prospects, including the Company's views with respect to the potential for mRNA and other pipeline therapeutics; BioNTech's efforts to combat COVID-19; the collaborations between BioNTech and Pfizer and Fosun to develop a potential COVID-19 vaccine; our expectations regarding the potential characteristics of BNT162b2 in our continuing Phase 2/3 trial and/or in commercial use based on data observations to date; the expected timepoint for additional readouts on efficacy data of BNT162b2 in our Phase 2/3 trial; the nature of the clinical data for BNT162, BNT311 and our other product candidates, which is subject to ongoing peer review, regulatory review and market interpretation; the timing for submission of data for, or receipt of, any potential approval or Emergency Use Authorization with respect to our BNT162 program; the timing for submission of BNT162 manufacturing data to the FDA; the ability of BioNTech to supply the quantities of BNT162 to support clinical development and, if approved, market demand, including our production estimates for 2020 and 2021 and orders received to-date; the planned next steps in BioNTech's pipeline programs and specifically including, but not limited to, statements regarding plans to initiate clinical trials of BioNTech's product candidates and expectations for data announcements with -
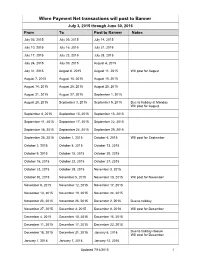
Transactions Posted to Pathway
When Payment Net transactions will post to Banner July 3, 2015 through June 30, 2016 From To Post to Banner Notes July 03, 2015 July 09, 2015 July 14, 2015 July 10, 2015 July 16, 2015 July 21, 2015 July 17, 2015 July 23, 2015 July 28, 2015 July 24, 2015 July 30, 2015 August 4, 2015 July 31, 2015 August 6, 2015 August 11, 2015 Will post for August August 7, 2015 August 13, 2015 August 18, 2015 August 14, 2015 August 20, 2015 August 25, 2015 August 21, 2015 August 27, 2015 September 1, 2015 August 28, 2015 September 3, 2015 September 9, 2015 Due to holiday at Monday Will post for August September 4, 2015 September 10, 2015 September 15, 2015 September 11, 2015 September 17, 2015 September 22, 2015 September 18, 2015 September 24, 2015 September 29, 2015 September 25, 2015 October 1, 2015 October 6, 2015 Will post for September October 2, 2015 October 8, 2015 October 13, 2015 October 9, 2015 October 15, 2015 October 20, 2015 October 16, 2015 October 22, 2015 October 27, 2015 October 23, 2015 October 29, 2015 November 3, 2015 October 30, 2015 November 5, 2015 November 10, 2015 Will post for November November 6, 2015 November 12, 2015 November 17, 2015 November 13, 2015 November 19, 2015 November 24, 2015 November 20, 2015 November 26, 2015 December 2, 2015 Due to holiday November 27, 2015 December 3, 2015 December 8, 2018 Will post for December December 4, 2015 December 10, 2015 December 15, 2015 December 11, 2015 December 17, 2015 December 22, 2015 December 18, 2015 December 31, 2015 January 6, 2016 Due to holiday closure Will post for December -

Outcome Reporting Bias in COVID-19 Mrna Vaccine Clinical Trials
medicina Perspective Outcome Reporting Bias in COVID-19 mRNA Vaccine Clinical Trials Ronald B. Brown School of Public Health and Health Systems, University of Waterloo, Waterloo, ON N2L3G1, Canada; [email protected] Abstract: Relative risk reduction and absolute risk reduction measures in the evaluation of clinical trial data are poorly understood by health professionals and the public. The absence of reported absolute risk reduction in COVID-19 vaccine clinical trials can lead to outcome reporting bias that affects the interpretation of vaccine efficacy. The present article uses clinical epidemiologic tools to critically appraise reports of efficacy in Pfzier/BioNTech and Moderna COVID-19 mRNA vaccine clinical trials. Based on data reported by the manufacturer for Pfzier/BioNTech vaccine BNT162b2, this critical appraisal shows: relative risk reduction, 95.1%; 95% CI, 90.0% to 97.6%; p = 0.016; absolute risk reduction, 0.7%; 95% CI, 0.59% to 0.83%; p < 0.000. For the Moderna vaccine mRNA-1273, the appraisal shows: relative risk reduction, 94.1%; 95% CI, 89.1% to 96.8%; p = 0.004; absolute risk reduction, 1.1%; 95% CI, 0.97% to 1.32%; p < 0.000. Unreported absolute risk reduction measures of 0.7% and 1.1% for the Pfzier/BioNTech and Moderna vaccines, respectively, are very much lower than the reported relative risk reduction measures. Reporting absolute risk reduction measures is essential to prevent outcome reporting bias in evaluation of COVID-19 vaccine efficacy. Keywords: mRNA vaccine; COVID-19 vaccine; vaccine efficacy; relative risk reduction; absolute risk reduction; number needed to vaccinate; outcome reporting bias; clinical epidemiology; critical appraisal; evidence-based medicine Citation: Brown, R.B.