Kidney Disease Caused by Dysregulation of the Complement Alternative Pathway: an Etiologic Approach
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Selected Candidates for 12Th Batch.Pdf
Punjab Security Training Institute, Jahan Khelan Selected Candidates for 12th Basic Security Training Course Commencing W.E.F 31.07.2011 Sr. ID Candidate Father's Mother's Address District No. No. Name Name Name 1 1 Jagseer Singh Manjeet Singh Rajinder Kaur Vpo. Kanjla, Tehsil Dhuri Sangrur 2 2 Karnail Singh Bhura Singh Amarjit Kaur VPO. Loha khera Sangrur 3 3 Sharanjeet Magh Singh Gurmail Kaur # 120. ward no. 11A, Moh. Sangrur Singh Shivpuri, near FCI Godown, Dhuri, Tehsil. Dhuri 4 4 Harjinder Tara Singh Baljinder Vill. Belewal (Darogewal) Po. Sangrur Singh Kaur Banbhora Tehsil Dhuri 5 5 Parminder Kulwinder Harminder Ramnagar W.no.5 Sunam Sangrur Singh Singh Kaur 6 6 Jasvir Singh Shingara Manjeet Kaur H.no.14,VPO. Loha khera Sangrur Singh 7 7 Jagseer Singh Sudagar Paramjeet VPO. Loha khera Sangrur Singh Kaur 8 8 Tejinder Singh Kewal Singh Manpreet # 269, ward no-8A, Gali no-2, Sangrur Kaur Guru Teg Bahadur Nagar, Dhuri, Tehsil. Dhuri 9 9 Rajpal Singh Hardial Singh Manjeet Kaur Vill.Issi Tehsil Dhuri Sangrur 10 11 Sukhchain Roop Singh Dhan Kaur Vill. Khanal Kalan, Teshil Sangrur Singh Sunam 11 12 Sultan Bashir Shakeela Vill.Dehleez Kalan, PO. & Sangrur Mohmad Mohmad tehsil Maler Kotla 12 13 Gurdeep Bhadur Singh Paramjeet Amar colony, Malerkotla Sangrur Singh Kaur 13 14 Gagandeep Gurdev Singh Maya Kaur Vill. Alal Tehsil Dhuri Sangrur Singh 14 15 Baghel Singh Mohinder Sawarn Kaur VPO. Sherpur, Sangrur Singh 15 18 Jasveer Singh Mohinder Satya Devi Vill. Shergarh, Po. Dudian, Sangrur Singh Tehsil Moonak 16 19 Jagseer Singh Gurmail Singh Rachhpal Kaur Vill.Shergarh Singh Wala, Sangrur PO. -

1 ADVANCE CAUSE LIST of CASES (APPLT. SIDE) Meeting Link: Https
08.09.2021 1 ADVANCE CAUSE LIST OF CASES (APPLT. SIDE) MATTERS LISTED FOR 08.09.2021 COURT NO. 1 DIVISION BENCH-I HON'BLE THE CHIEF JUSTICE HON'BLE MS.JUSTICE JYOTI SINGH (PHYSICAL HEARING COURT) [NOTE: IN CASE ANY ASSISTANCE REGARDING VIRTUAL HEARING IS REQUIRED, PLEASE CONTACT, MR. VIJAY RATTAN SUNDRIYAL (PH: 9811136589) COURT MASTER & MR. VISHAL (PH 9968315312) ASSISTANT COURT MASTER TO HON'BLE THE CHIEF JUSTICE & PLEASE CONTACT, MR.SANDEEP (9810713157) COURT MASTER AND MR.AJAY KUMAR MAVI,ACM (PH:-9968303497) TO HON’BLE MS. JUSTICE JYOTI SINGH.] [NOTE: ALL COUNSEL/PARTIES ARE REQUESTED TO KEEP THEIR WEB CAMERA OFF AND MIC MUTED UNLESS THEIR MATTER IS GOING ON] Meeting link: https://dhcvirtualcourt.webex.com/dhcvirtualcourt/j.php?MTID=m3f5985f2f8a66575a1b005de249eab82 Meeting number :- 2515 996 4520 Password :- 1234 FRESH MATTERS & APPLICATIONS ______________________________ 1. W.P.(C) 4917/2021 FRESH FRUIT FLOWERS AND HIMANSHU GARG,RIPUDAMAN VEGETABLES TRADERS SINGH BHARDWAJ ASSOCIATION Vs. DIRECTORATE GENERAL OF FOREIGN TRADE & ANR. ADVOCATE DETAILS FOR ABOVE CASE shakeel sarwar wani and himanshu garg()(8750618722)(Petitioner) HIMANSHUGARG([email protected])(9654542581)(Petitioner) mohammad muqeem([email protected])(9999864964)(Respondent) FRESH FRUIT FLOWERS AND VEGETABLES TRADERS ASSOCIATION([email protected])()(Respondent) FRESH FRUIT FLOWERS AND VEGETABLES TRADERS ASSOCIATION([email protected])()(Respondent) DEPARTMENT OF ARGICULTURE, COOPERATION AND FARMERS WELFARE([email protected])()(Respondent) DIRECTORATE GENERAL OF FOREIGN TRADE([email protected])()(Respondent) DEPARTMENT OF ARGICULTURE, COOPERATION AND FARMERS WELFARE([email protected])()(Respondent) DIRECTORATE GENERAL OF FOREIGN TRADE([email protected])()(Respondent) ripu daman bhardwaj()(9818030700)(Petitioner) s s wani associates([email protected])()(Respondent) 2. -

Punjab Caste-System and Voting Behavior
Punjab Caste-System and Voting Behavior * Syed Karim Haider Abstract This article examines the impact of Punjab‟s caste-system on voting behavior emerging from 2013 Pakistan‟s general elections. Historically, the caste and Biradari system have been playing a significant role in the region of the Punjab since the arrival of Aryans. The historical evolution and modernization of the British Raj never fully changed the traditional value-system of the Punjabi society which is based on caste and biradari. With particular reference to caste and Biradari system, an analytical study has been made to understand deep roots of Caste-System and its impact on the voting behavior of four selected districts of the province of Punjab in 2013 general elections of Pakistan. Further, this research shows that the Punjabi society is based on multiculturalism and social diversification with parochial political culture; therefore, the Punjabi society accepts authoritarian rule which begins from family and ends at national politics. Introduction The Province of Punjab has been considered an important region of the subcontinent from very beginning. Geographically, it is located on the North and Western border of the subcontinent and historically it has a significance which goes back to the Indus civilization. Due to its geography and history, it has been an important region which continuously remained a target of foreign invaders. As a result, the province of Punjab has remained under the great influence of multiculturalism and social diversification which resulted in the development of caste-based socio-political and cultural system which has motivated the author to analyze the political behavior of the Punjab, which is deeply rooted in the caste and biradari system. -

Singapore's Early Sikh Pioneers
SINGAPORE’S EARLY SIKH PIONEERS Origins, Settlement, Contributions and Institutions RISHPAL SINGH SIDHU CENTRAL SIKH GURDWARA BOARD SINGAPORE Singapore’s Early Sikh Pioneers: Origins, Settlement, Contributions and Institutions Rishpal Singh Sidhu Compiler & Editor CENTRAL SIKH GURDWARA BOARD SINGAPORE Front Cover Photo: A collage of the seven Sikh Gurdwaras and Singapore Khalsa Association in Singapore Back Cover Photo: A collage of some of Singapore’s Early Sikh Pioneers Copyright, Central Sikh Gurdwara Board, Singapore, 2017 ISBN: 978-981-09-4437-7 Printed by: Khalsa Printers Pte Ltd, Singapore DEDICATION Dedicated to Sikh youth in Singapore in the fervent belief they will build on the achievements and contributions of their forebears for a better and brighter tomorrow. OUR SPONSOR Central Sikh Gurdwara Board would like to express their heartfelt thanks to our Patron, S. Naranjan Singh Brahmpura for sponsoring the cost of publishing this book. Naranjan Singh Brahmpura Patron Central Sikh Gurdwara Board Singapore Khalsa Association Trustee Singapore Sikh Education Foundation Sikh Welfare Council Past President Central Sikh Gurdwara Board Sri Guru Singh Sabha CONTENTS Foreword 6 Preface 7 Acknowledgements 8 Fast forward 9 1 Introduction 11 2 Singapore’s first Sikh 15 3 Sikh migration to Singapore: Phases and patterns 21 4 Early Sikh settlers in Singapore 31 5 Sikhs in the British Naval Base 39 6 Establishment of Gurdwaras, Sikh Advisory Board and other Sikh institutions 43 7 Sikh soldiers involvement in the defense of Singapore in World War II and civilian life during the Japanese Occupation 97 8 Early Sikh pioneers and their contributions to nation building 109 9 Colonial Singapore’s first Sikh politician 155 10. -
FINAL DISTRIBUTION.Xlsx
Annexure-1A 1)Taxpayers with turnover above Rs 1.5 Crores a) Taxpayers falling under the jurisdiction of the Centre Taxpayer's Name SL NO GSTIN Registration Name TRADE_NAME 1 EASTERN COAL FIELDS LTD. EASTERN COAL FIELDS LTD. 19AAACE7590E1ZI 2 SAIL (D.S.P) SAIL (D.S.P) 19AAACS7062F6Z6 3 CESC LTD. CESC LIMITED 19AABCC2903N1ZL 4 MATERIALS CHEMICALS AND PERFORMANCE INTERMEDIARIESMCC PTA PRIVATE INDIA CORP.LIMITED PRIVATE LIMITED 19AAACM9169K1ZU 5 N T P C / F S T P P LIMITED N T P C / F S T P P LIMITED 19AAACN0255D1ZV 6 DAMODAR VALLEY CORPORATION DAMODAR VALLEY CORPORATION 19AABCD0541M1ZO 7 BANK OF NOVA SCOTIA 19AAACB1536H1ZX 8 DHUNSERI PETGLOBAL LIMITED DHUNSERI PETGLOBAL LIMITED 19AAFCD5214M1ZG 9 E M C LTD 19AAACE7582J1Z7 10 BHARAT SANCHAR NIGAM LIMITED BHARAT SANCHAR NIGAM LIMITED 19AABCB5576G3ZG 11 HINDUSTAN UNILEVER LIMITED 19AAACH1004N1ZR 12 GUJARAT COOPERATIVE MILKS MARKETING FEDARATION LTD 19AAAAG5588Q1ZT 13 VODAFONE MOBILE SERVICES LIMITED VODAFONE MOBILE SERVICES LIMITED 19AAACS4457Q1ZN 14 N MADHU BHARAT HEAVY ELECTRICALS LTD 19AAACB4146P1ZC 15 JINDAL INDIA LTD 19AAACJ2054J1ZL 16 SUBRATA TALUKDAR HALDIA ENERGY LIMITED 19AABCR2530A1ZY 17 ULTRATECH CEMENT LIMITED 19AAACL6442L1Z7 18 BENGAL ENERGY LIMITED 19AADCB1581F1ZT 19 ANIL KUMAR JAIN CONCAST STEEL & POWER LTD.. 19AAHCS8656C1Z0 20 ELECTROSTEEL CASTINGS LTD 19AAACE4975B1ZP 21 J THOMAS & CO PVT LTD 19AABCJ2851Q1Z1 22 SKIPPER LTD. SKIPPER LTD. 19AADCS7272A1ZE 23 RASHMI METALIKS LTD 19AACCR7183E1Z6 24 KAIRA DISTRICT CO-OP MILK PRO.UNION LTD. KAIRA DISTRICT CO-OP MILK PRO.UNION LTD. 19AAAAK8694F2Z6 25 JAI BALAJI INDUSTRIES LIMITED JAI BALAJI INDUSTRIES LIMITED 19AAACJ7961J1Z3 26 SENCO GOLD LTD. 19AADCS6985J1ZL 27 PAWAN KR. AGARWAL SHYAM SEL & POWER LTD. 19AAECS9421J1ZZ 28 GYANESH CHAUDHARY VIKRAM SOLAR PRIVATE LIMITED 19AABCI5168D1ZL 29 KARUNA MANAGEMENT SERVICES LIMITED 19AABCK1666L1Z7 30 SHIVANANDAN TOSHNIWAL AMBUJA CEMENTS LIMITED 19AAACG0569P1Z4 31 SHALIMAR HATCHERIES LIMITED SHALIMAR HATCHERIES LTD 19AADCS6537J1ZX 32 FIDDLE IRON & STEEL PVT. -

2021 Daily Prayer Guide for All People Groups & LR-Unreached People Groups = LR-Upgs
2021 Daily Prayer Guide for all People Groups & LR-Unreached People Groups = LR-UPGs - of INDIA Source: Joshua Project data, www.joshuaproject.net Western edition To order prayer resources or for inquiries, contact email: [email protected] I give credit & thanks to Create International for permission to use their PG photos. 2021 Daily Prayer Guide for all People Groups & LR-UPGs = Least-Reached-Unreached People Groups of India INDIA SUMMARY: 2,717 total People Groups; 2,445 LR-UPG India has 1/3 of all UPGs in the world; the most of any country LR-UPG definition: 2% or less Evangelical & 5% or less Christian Frontier (FR) definition: 0% to 0.1% Christian Why pray--God loves lost: world UPGs = 7,407; Frontier = 5,042. Color code: green = begin new area; blue = begin new country Downloaded from www.joshuaproject.net in September 2020 * * * "Prayer is not the only thing we can can do, but it is the most important thing we can do!" * * * India ISO codes are used for some Indian states as follows: AN = Andeman & Nicobar. JH = Jharkhand OD = Odisha AP = Andhra Pradesh+Telangana JK = Jammu & Kashmir PB = Punjab AR = Arunachal Pradesh KA = Karnataka RJ = Rajasthan AS = Assam KL = Kerala SK = Sikkim BR = Bihar ML = Meghalaya TN = Tamil Nadu CT = Chhattisgarh MH = Maharashtra TR = Tripura DL = Delhi MN = Manipur UT = Uttarakhand GJ = Gujarat MP = Madhya Pradesh UP = Uttar Pradesh HP = Himachal Pradesh MZ = Mizoram WB = West Bengal HR = Haryana NL = Nagaland Why Should We Pray For Unreached People Groups? * Missions & salvation of all people is God's plan, God's will, God's heart, God's dream, Gen. -

Find Address of Your Nearest Loan Center and Phone Number of Concerned Focal Person
Find address of your nearest loan center and phone number of concerned focal person Loan Center/ S.No. Province District PO Name City / Tehsil Focal Person Contact No. Union Council/ Location Address Branch Name Akhuwat Islamic College Chowk Oppsite Boys College 1 Azad Jammu and Kashmir Bagh Bagh Bagh Nadeem Ahmed 0314-5273451 Microfinance (AIM) Sudan Galli Road Baagh Akhuwat Islamic Muzaffarabad Road Near main bazar 2 Azad Jammu and Kashmir Bagh Dhir Kot Dhir Kot Nadeem Ahmed 0314-5273451 Microfinance (AIM) dhir kot Akhuwat Islamic Mang bajri arja near chambar hotel 3 Azad Jammu and Kashmir Bagh Harighel Harighel Nadeem Ahmed 0314-5273451 Microfinance (AIM) Harighel Akhuwat Islamic 4 Azad Jammu and Kashmir Bhimber Bhimber Bhimber Arshad Mehmood 0346-4663605 Kotli Mor Near Muslim & School Microfinance (AIM) Akhuwat Islamic 5 Azad Jammu and Kashmir Bhimber Barnala Barnala Arshad Mehmood 0346-4663605 Main Road Bimber & Barnala Road Microfinance (AIM) Akhuwat Islamic Main choki Bazar near Sir Syed girls 6 Azad Jammu and Kashmir Bhimber Samahni Samahni Arshad Mehmood 0346-4663605 Microfinance (AIM) College choki Samahni Helping Hand for Adnan Anwar HHRD Distrcict Office Relief and Hattian,Near Smart Electronics,Choke 7 Azad Jammu and Kashmir Hattian Hattian UC Hattian Adnan Anwer 0341-9488995 Development Bazar, PO, Tehsil and District (HHRD) Hattianbala. Helping Hand for Adnan Anwar HHRD Distrcict Office Relief and Hattian,Near Smart Electronics,Choke 8 Azad Jammu and Kashmir Hattian Hattian UC Langla Adnan Anwer 0341-9488995 Development Bazar, PO, Tehsil and District (HHRD) Hattianbala. Helping Hand for Relief and Zahid Hussain HHRD Lamnian office 9 Azad Jammu and Kashmir Hattian Hattian UC Lamnian Zahid Hussain 0345-9071063 Development Main Lamnian Bazar Hattian Bala. -

Pioneer Voices from California Reflections on Race, Religion, & Ethnicity
5 KAREN LEONARD Pioneer Voices from California Reflections on Race, Religion, & Ethnicity It has become common to talk of the Sikh diaspora, but there is some question whether or not "Sikh'' is the most appropriate category for analysis of these emigrants fro~ South Asia. While the overwhelming majority of the Punjabi pioneers in early twentieth century Qilifomia were indeed Sikhs, my .research indicates that religion was less salient than other characteristics for these men. It was in fact a Punjabi diaspora, and to go back and emphasize Sikhs and Sikhism does violence to the historical experiences of the immigrant<; and their descendants. Most research on the Punjabi immigrants has focused on their public life. It is clear that these pioneers in the American West contended with racial, religious, and ethnic prejudices all their lives in the U.S. They battled for citizenship and lost; losing that, they fell prey to the Alien Land Laws of California and Arizona and could continue farming only in ways that put them at the mercy of non-Indians; theirGhadar party activities and conflicts with each other were magnified and misinterpreted in the press. Scholars have recently clarified, corrected, and enlarged upon the early materials about the public life of the men from the · Punjab (Barrie~, this volume; Jacoby, 1978; Puri, 1980). Pioneer Vo~es From California 121 It is equally important to look at the immigrants' private lives, at family life, where U.S. immigration prohibitions and California miscegenation lawsdictatedan overwhelmingly non Punjabi and, indeed, an Hispanic origin for the women with whom most Punjabi men formed families.• Men and women applying to the County Clerk for a marriage license had to be of the same race, had to look alike, and most often it was Hispanic women who satisfied that requirement. -
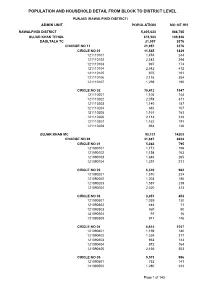
Rawalpindi Blockwise
POPULATION AND HOUSEHOLD DETAIL FROM BLOCK TO DISTRICT LEVEL PUNJAB (RAWALPINDI DISTRICT) ADMIN UNIT POPULATION NO OF HH RAWALPINDI DISTRICT 5,405,633 888,765 GUJAR KHAN TEHSIL 678,503 109,826 DAULTALA TC 21,957 3376 CHARGE NO 11 21,957 3376 CIRCLE NO 01 11,545 1829 121110101 1,474 244 121110102 2,143 294 121110103 997 174 121110104 2,542 412 121110105 975 161 121110106 2,116 354 121110107 1,298 190 CIRCLE NO 02 10,412 1547 121110201 1,105 144 121110202 2,078 311 121110203 1,140 187 121110204 682 107 121110205 1,167 163 121110206 2,114 318 121110207 1,162 191 121110208 964 126 GUJAR KHAN MC 90,131 14203 CHARGE NO 09 41,687 6634 CIRCLE NO 01 5,363 795 121090101 1,172 156 121090102 1,154 163 121090103 1,646 265 121090104 1,391 211 CIRCLE NO 02 6,320 962 121090201 1,510 224 121090202 1,203 186 121090203 1,587 239 121090204 2,020 313 CIRCLE NO 03 3,051 453 121090301 1,039 130 121090302 444 71 121090303 560 90 121090304 97 16 121090305 911 146 CIRCLE NO 04 6,614 1057 121090401 1,198 180 121090402 1,324 217 121090403 963 143 121090404 973 164 121090405 2,156 353 CIRCLE NO 05 5,573 996 121090501 752 141 121090502 1,280 233 Page 1 of 143 POPULATION AND HOUSEHOLD DETAIL FROM BLOCK TO DISTRICT LEVEL PUNJAB (RAWALPINDI DISTRICT) ADMIN UNIT POPULATION NO OF HH 121090503 1,083 196 121090504 1,313 233 121090505 1,145 193 CIRCLE NO 06 5,306 839 121090601 774 117 121090602 1,193 193 121090603 832 143 121090604 1,634 244 121090605 873 142 CIRCLE NO 07 5,736 921 121090701 1,128 178 121090702 547 104 121090703 1,670 261 121090704 2,391 378 CIRCLE NO 08 -

Rollno Name Father's Name Marks 240001 MANPREET KAUR SURJIT SINGH 39 240002 GAGANDEEP KAUR KULDIP SINGH 39 240003 MEENAKSHI KHAY
Baba Farid University of Health Sciences,Faridkot Provisional Result of MPHW(Female) test dated 26-10-2020 Rollno Name Father's name Marks 240001 MANPREET KAUR SURJIT SINGH 39 240002 GAGANDEEP KAUR KULDIP SINGH 39 240003 MEENAKSHI KHAYALI RAM 47 240006 RUPI RANI KARAM CHAND 46 240007 MANPREET KAUR SURINDER SINGH 34 240009 NAVNEET KAUR NIRMAL SINGH 34 240011 AMANJOT KAUR SUKHVIR SINGH 49 240013 SAVEENA KUMARI SHANKAR SINGH 42 240015 JYOTI LAL CHAND 32.75 240016 ARTI CHAUHAN BALWINDER SINGH 52 240017 SUMITA KUMARI SHANKAR SINGH 49 240019 TARVINDER KAUR GURBAX SINGH 48 240020 MANPREET KAUR DILBAG SINGH 41 240021 SUNEET KAUR HARDEEP SINGH 41 240022 JAGDEEP KAUR SHAMSHER SINGH 71 240023 NARINDER KAUR RAJ KUMAR 30.75 240026 NAZRUN NISHA HASMULLA ANSARI 52 240027 BABITA SHARMA BAM NATH 38 240028 CHANCHAL RANI RAMPAL 55 240029 RAJ RANI RAKHA RAM 40 240030 PREETI CHOUDHARY BALRAM 47 240031 HARPREET KAUR GURMEET SINGH 33 240032 SHAMA DEVI JIYA LAL 49 240033 SHABNAM JARNAIL SINGH 30 240034 SUMAN DEVI DARSHAN KUMAR 26.25 240036 BALJEET KAUR AMARJEET SINGH 55 240037 MANJIT KAUR HARBANS SINGH 50 240038 RAJIA SULTANA MURAD ALI 38 240039 MANPREET KAUR JARNAIL SINGH 55 240040 SHEEL KUMARI MADAN LAL 47 240041 SANDEEP KAUR KAMAL DEV 29 240042 RAJBIR KAUR GURCHARAN SINGH 57 240043 MEENAKASHI DEVI HUSAN LAL 45 240045 RAMANDEEP KAUR DHANA SINGH 44 240047 RAJWINDER KAUR HARJAP SINGH 45 240048 NISHA RANI SUKHWINDER SINGH 31 240049 SARITA SHARMA PREM CHAND SHARMA 44 240050 KULWINDER KAUR RATTAN SINGH 35 Baba Farid University of Health Sciences,Faridkot Provisional -

Jarnail Singh Bhindranwale: a Charismatic Authority and His Ideology John P
Florida International University FIU Digital Commons FIU Electronic Theses and Dissertations University Graduate School 3-22-2017 Jarnail Singh Bhindranwale: A Charismatic Authority and His Ideology John P. Cibotti Florida International University, [email protected] DOI: 10.25148/etd.FIDC001784 Follow this and additional works at: https://digitalcommons.fiu.edu/etd Part of the Anthropological Linguistics and Sociolinguistics Commons, Asian Studies Commons, Child Psychology Commons, Community Psychology Commons, Human Ecology Commons, Human Geography Commons, Political Economy Commons, Politics and Social Change Commons, Race and Ethnicity Commons, Regional Sociology Commons, Social and Cultural Anthropology Commons, Social Psychology Commons, Sociology of Culture Commons, and the Sociology of Religion Commons Recommended Citation Cibotti, John P., "Jarnail Singh Bhindranwale: A Charismatic Authority and His Ideology" (2017). FIU Electronic Theses and Dissertations. 3190. https://digitalcommons.fiu.edu/etd/3190 This work is brought to you for free and open access by the University Graduate School at FIU Digital Commons. It has been accepted for inclusion in FIU Electronic Theses and Dissertations by an authorized administrator of FIU Digital Commons. For more information, please contact [email protected]. FLORIDA INTERNATIONAL UNIVERSITY Miami, Florida JARNAIL SINGH BHINDRANWALE: A CHARISMATIC AUTHORITY AND HIS IDEOLOGY A thesis submitted in partial fulfillment of the requirements for the degree of MASTER OF ARTS in RELIGIOUS STUDIES by John Paul Cibotti 2017 To: Dean John F. Stack Steven J. Green School of International and Public Affairs This thesis, written by John Paul Cibotti, and entitled Jarnail Singh Bhindranwale: A Charismatic Authority and His Ideology, having been approved in respect to style and intellectual content, is referred to you for judgment. -

YPS Yearbook 2013-14.Pdf
School Motto Vidya : Education What sculpture is to a block of marble, education is to the human soul - Addison Vinay : Humility The wiser you are, the more you realize how little you actually know - Socrates Veerta : Courage Courage is not simply one of the virtues but the form of every virtue at the testing point - C.S. Lewis 00 CONTENTS 04 30 88 92 Principal's Message SCHOOL REPORTS REACHING OUT HOUSE REPORTS Editorial Kindergarten Building Bridges Aitchison House Prefect's Council Junior School Achievers Beyond School Nalagarh House ISC 2014 Senior School Patiala House ICSE 2014 Ranjit House Academic Staff Tagore House Administrative Staff Boarding House Support Staff Investiture Ceremony Founder's Day YPS Xtravaganza Celebrations 114 Farewell Class XII Bidding Adieu PRINCIPAL’S MESSAGE After six years in YPS Patiala, and the parents, students and staff. four years with an International This is a time for change for IB school in Bali, Indonesia, I am everyone and for me as well. extremely delighted to be back and to be a part of the YPS MUNs, Treks, the annual play community again. YPS Mohali has 'Anne of Green Gables', Sports dedicated and talented staff and Day, Debates...all this and more students, and together the YPS recorded in the pages of this family can only grow from edition of the yearbook. The strength to strength. I am Editorial team has done a overwhelmed and honoured by wonderful job bringing out the the support I have received from yearbook for 2013. This is a 04 book of memories...of the out of the campus throughout written word, and of visuals the year.