Transcription Start Site Analysis Reveals Widespread Divergent Transcription in D. Melanogaster and Core Promoter-Encoded Enhanc
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mutations in DCPS and EDC3 in Autosomal Recessive Intellectual
Human Molecular Genetics, 2015, Vol. 24, No. 11 3172–3180 doi: 10.1093/hmg/ddv069 Advance Access Publication Date: 20 February 2015 Original Article Downloaded from ORIGINAL ARTICLE Mutations in DCPS and EDC3 in autosomal recessive intellectual disability indicate a crucial role for mRNA http://hmg.oxfordjournals.org/ decapping in neurodevelopment Iltaf Ahmed1,2,†, Rebecca Buchert3,†, Mi Zhou5,†, Xinfu Jiao5,†, Kirti Mittal1, Taimoor I. Sheikh1, Ute Scheller3, Nasim Vasli1, Muhammad Arshad Rafiq1, 6 1 7 2 M. Qasim Brohi , Anna Mikhailov , Muhammad Ayaz , Attya Bhatti , at Universitaet Erlangen-Nuernberg, Wirtschafts- und Sozialwissenschaftliche Z on August 15, 2016 Heinrich Sticht4, Tanveer Nasr8,9, Melissa T. Carter10, Steffen Uebe3, André Reis3, Muhammad Ayub7,11, Peter John2, Megerditch Kiledjian5,*, John B. Vincent1,12,13,* and Rami Abou Jamra3,* 1Molecular Neuropsychiatry and Development Lab, Campbell Family Mental Health Research Institute, Centre for Addiction and Mental Health, 250 College Street, Toronto, Ontario, Canada M5T 1R8, 2Atta-ur-Rehman School of Applied Biosciences (ASAB), National University of Sciences and Technology (NUST), Islamabad 44000, Pakistan, 3Institute of Human Genetics and 4Bioinformatics, Institute of Biochemistry, Friedrich-Alexander-Universität Erlangen-Nürnberg, Erlangen 91054, Germany, 5Department of Cell Biology and Neuroscience, Rutgers University, Piscataway, NJ 08854, USA, 6Sir Cowasji Jehangir Institute of Psychiatry, Hyderabad, Sindh 71000, Pakistan, 7Lahore Institute of Research and Development, -

Dcps Is a Transcript-Specific Modulator of RNA in Mammalian Cells
Downloaded from rnajournal.cshlp.org on September 28, 2021 - Published by Cold Spring Harbor Laboratory Press DcpS is a transcript-specific modulator of RNA in mammalian cells MI ZHOU,1 SOPHIE BAIL,1 HEATHER L. PLASTERER,2 JAMES RUSCHE,2 and MEGERDITCH KILEDJIAN1 1Department of Cell Biology and Neuroscience, Rutgers University, Piscataway, New Jersey 08854, USA 2Repligen Corporation, Waltham, Massachusetts 02453, USA ABSTRACT The scavenger decapping enzyme DcpS is a multifunctional protein initially identified by its property to hydrolyze the resulting cap structure following 3′ end mRNA decay. In Saccharomyces cerevisiae, the DcpS homolog Dcs1 is an obligate cofactor for the 5′-3′ exoribonuclease Xrn1 while the Caenorhabditis elegans homolog Dcs-1, facilitates Xrn1 mediated microRNA turnover. In both cases, this function is independent of the decapping activity. Whether DcpS and its decapping activity can affect mRNA steady state or stability in mammalian cells remains unknown. We sought to determine DcpS target genes in mammalian cells using a cell-permeable DcpS inhibitor compound, RG3039 initially developed for therapeutic treatment of spinal muscular atrophy. Global mRNA levels were examined following DcpS decapping inhibition with RG3039. The steady-state levels of 222 RNAs were altered upon RG3039 treatment. Of a subset selected for validation, two transcripts that appear to be long noncoding RNAs HS370762 and BC011766, were dependent on DcpS and its scavenger decapping catalytic activity and referred to as DcpS-responsive noncoding transcripts (DRNT) 1 and 2, respectively. Interestingly, only the increase in DRNT1 transcript was accompanied with an increase of its RNA stability and this increase was dependent on both DcpS and Xrn1. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Identification of Enhancer of Mrna Decapping 4 As a Novel Fusion Partner of MLL in Acute Myeloid Leukemia
STIMULUS REPORT Identification of enhancer of mRNA decapping 4 as a novel fusion partner of MLL in acute myeloid leukemia Heiko Becker,1-3 Gabriele Greve,1,2 Keisuke Kataoka,4 Jan-Philipp Mallm,5,6 Jesus´ Duque-Afonso,1,2,7 Tobias Ma,1,2 Christoph Niemoller,¨ 1,2 Milena Pantic,1 Justus Duyster,1-3 Michael L. Cleary,7 Julia Schuler,¨ 8 Karsten Rippe,5,6 Seishi Ogawa,3 and Michael Lubbert¨ 1-3 1Department of Medicine I, Medical Center, and 2Faculty of Medicine, University of Freiburg, Freiburg, Germany; 3German Cancer Consortium partner site, Freiburg, Germany; 4Department of Pathology and Tumor Biology, Kyoto University, Kyoto, Japan; 5Division of Chromatin Networks and 6Single-cell Open Laboratory, German Cancer Research Center, Heidelberg, Germany; 7Department of Pathology, Stanford University, Stanford, CA; and 8Charles River Discovery Research Services Germany GmbH, Freiburg, Germany Key Points Introduction • mRNA decapping gene Translocations involving MLL (aka KMT2A) located on chromosome 11q23 occur in acute myeloid EDC4 is a novel fusion leukemia (AML) and lymphoblastic leukemia. In AML, they generally confer an adverse prognosis, unless MLL partner of in AML. the MLLT3 (aka AF9) gene is involved.1 More than 130 different translocation partner genes (TPGs) MLL 2 • Genes functioning in have been identified, forming the recombinome. mRNA decapping may Recently, the scavenger messenger RNA (mRNA) decapping enzyme DCPS has been identified to be compose a distinct required for survival of AML cells, but not normal hematopoietic cells, and a DCPS inhibitor showed antileukemic activity.3,4 DCPS is also 1 of 2 genes (the other being DCP1A) involved in mRNA group of MLL fusion decapping and having been described as TPG of MLL in single leukemia cases.5-7 partners that links MLL MLL function with mRNA Here, we describe a novel fusion with another mRNA decapping component, ie, the enhancer of mRNA decapping 4 gene (EDC4;alsoknownasGE1 or HEDLS), in AML. -
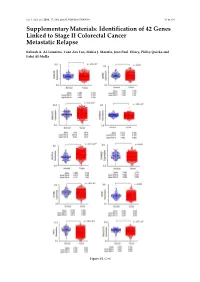
Identification of 42 Genes Linked to Stage II Colorectal Cancer Metastatic Relapse
Int. J. Mol. Sci. 2016, 17, 598; doi:10.3390/ijms17040598 S1 of S16 Supplementary Materials: Identification of 42 Genes Linked to Stage II Colorectal Cancer Metastatic Relapse Rabeah A. Al-Temaimi, Tuan Zea Tan, Makia J. Marafie, Jean Paul Thiery, Philip Quirke and Fahd Al-Mulla Figure S1. Cont. Int. J. Mol. Sci. 2016, 17, 598; doi:10.3390/ijms17040598 S2 of S16 Figure S1. Mean expression levels of fourteen genes of significant association with CRC DFS and OS that are differentially expressed in normal colon compared to CRC tissues. Each dot represents a sample. Table S1. Copy number aberrations associated with poor disease-free survival and metastasis in early stage II CRC as predicted by STAC and SPPS combined methodologies with resident gene symbols. CN stands for copy number, whereas CNV is copy number variation. Region Cytoband % of CNV Count of Region Event Gene Symbols Length Location Overlap Genes chr1:113,025,076–113,199,133 174,057 p13.2 CN Loss 0.0 2 AKR7A2P1, SLC16A1 chr1:141,465,960–141,822,265 356,305 q12–q21.1 CN Gain 95.9 1 SRGAP2B MIR5087, LOC10013000 0, FLJ39739, LOC10028679 3, PPIAL4G, PPIAL4A, NBPF14, chr1:144,911,564–146,242,907 1,331,343 q21.1 CN Gain 99.6 16 NBPF15, NBPF16, PPIAL4E, NBPF16, PPIAL4D, PPIAL4F, LOC645166, LOC388692, FCGR1C chr1:177,209,428–177,226,812 17,384 q25.3 CN Gain 0.0 0 chr1:197,652,888–197,676,831 23,943 q32.1 CN Gain 0.0 1 KIF21B chr1:201,015,278–201,033,308 18,030 q32.1 CN Gain 0.0 1 PLEKHA6 chr1:201,289,154–201,298,247 9093 q32.1 CN Gain 0.0 0 chr1:216,820,186–217,043,421 223,235 q41 CN -

The Natural Antisense Transcript NATTD Regulates the Transcription
International Journal of Biochemistry and Cell Biology 110 (2019) 103–110 Contents lists available at ScienceDirect International Journal of Biochemistry and Cell Biology journal homepage: www.elsevier.com/locate/biocel The natural antisense transcript NATTD regulates the transcription of decapping scavenger (DcpS) enzyme T ⁎ Zhu-Zhong Mei , Hongwei Sun, Xiaoli Ou, Lei Li, Junwei Cai, Shuiwang Hu, Juan Wang, ⁎ ⁎ Haihua Luo, Jinghua Liu , Yong Jiang From Guangdong Provincial Key Laboratory of Proteomics, State Key Laboratory of Organ Failure Research, Department of Pathophysiology, Southern Medical University, Guangzhou 510515, China ARTICLE INFO ABSTRACT Keywords: Natural antisense transcripts (NATs) are transcribed from the opposite strand of other genes. Most of them are Natural antisense transcript noncoding RNAs. They have been reported to play important roles in a variety of biological processes. In this NATTD study, we identified a novel NAT, NATTD, which is partially complementary to both the TIRAP/Mal and DcpS DcpS genes. Interestingly, NATTD only positively regulates the expression of DcpS, a decapping scavenger enzyme Epigenetic regulation which is a promising therapeutic target for spinal muscular atrophy. But it has no obvious effects on the ex- pression of TIRAP/Mal gene. The NATTD transcript primarily resides in the nucleus and does not alter the mRNA stability of DcpS. Instead, it is required for the recruitment of RNA polymerase II at the mouse DcpS promoter. Chromatin immunoprecipitation assays revealed that knocking-down -

Role of Dcps in Mammalian RNA Regulation and Human Diseases
ROLE OF DCPS IN MAMMALIAN RNA REGULATION AND HUMAN DISEASES By MI ZHOU A dissertation submitted to the Graduate School-New Brunswick and The Graduate School of Biomedical Sciences Rutgers, The State University of New Jersey In partial fulfillment of the requirements For the degree of Doctor of Philosophy Graduate Program in Cell and Development Biology Written under the direction of Dr. Megerditch Kiledjian And approved by _________________________________ _________________________________ _________________________________ _________________________________ New Brunswick, New Jersey October, 2015 ABSTRACT OF THE DISSERTATION Role of DcpS in Mammalian RNA Regulation and Human Diseases By MI ZHOU Dissertation Director Dr. Megerditch Kiledjian In eukaryotic cells, mRNA degradation plays an important role in the control of gene expression and is therefore highly regulated. The scavenger decapping enzyme DcpS is a multifunctional protein that plays a critical role in mRNA degradation. We first sought to identify DcpS target genes in mammalian cells using a cell permeable DcpS inhibitor compound, RG3039, which was initially developed for therapeutic treatment of Spinal Muscular Atrophy (SMA). Microarray analysis following DcpS decapping inhibition by RG3039 revealed the steady state levels of 222 RNAs were altered. Of a subset selected for validation by qRT-PCR, two non-coding transcripts dependent on DcpS decapping activity, were identified and referred to as DcpS Responsive Noncoding Transcript (DRNT) 1 and 2 respectively. Only the increase in DRNT1 transcript was accompanied with an increase of its RNA stability and this increase was dependent on both DcpS and Xrn1. Our data indicate that DcpS is a transcript-restricted modulator of RNA stability in mammalian cells and the RG3039 ii quinazoline compound is pleotropic, influence gene expression in both an apparent DcpS dependent and independent manner. -

Ge-1 Is a Central Component of the Mammalian Cytoplasmic Mrna Processing Body
Ge-1 is a central component of the mammalian cytoplasmic mRNA processing body The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Yu, J. H. 2005. “Ge-1 Is a Central Component of the Mammalian Cytoplasmic mRNA Processing Body.” RNA 11 (12). Cold Spring Harbor Laboratory: 1795–1802. doi:10.1261/rna.2142405. Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:41555773 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA Downloaded from rnajournal.cshlp.org on October 16, 2019 - Published by Cold Spring Harbor Laboratory Press REPORT Ge-1 is a central component of the mammalian cytoplasmic mRNA processing body JIANG HONG YU,1,4 WEI-HONG YANG,1,4 TOD GULICK,2 KENNETH D. BLOCH,3 and DONALD B. BLOCH1 1Department of Medicine, Harvard Medical School, The Center for Immunology and Inflammatory Diseases; 2The Diabetes Research Laboratory; and 3The Cardiovascular Research Center of the General Medical Services, Massachusetts General Hospital, Boston, Massachusetts 02114, USA ABSTRACT The mRNA processing body (P-body) is a cellular structure that regulates gene expression by degrading cytoplasmic mRNA. The objective of this study was to identify and characterize novel components of the mammalian P-body. Approximately 5% of patients with the autoimmune disease primary biliary cirrhosis have antibodies directed against this structure. Serum from one of these patients was used to identify a cDNA encoding Ge-1, a 1401-amino-acid protein. -

The Dynamics of Mammalian P Body Transport, Assembly, and Disassembly in Vivo Adva Aizer,* Yehuda Brody,* Lian Wee Ler,† Nahum Sonenberg,† Robert H
Molecular Biology of the Cell Vol. 19, 4154–4166, October 2008 The Dynamics of Mammalian P Body Transport, Assembly, and Disassembly In Vivo Adva Aizer,* Yehuda Brody,* Lian Wee Ler,† Nahum Sonenberg,† Robert H. Singer,‡ and Yaron Shav-Tal* *The Mina and Everard Goodman Faculty of Life Sciences and Institute of Nanotechnology, Bar-Ilan University, Ramat Gan 52900, Israel; †Department of Biochemistry and McGill Cancer Center, McGill University, Montreal, Quebec H3G 1Y6, Canada; and ‡Department of Anatomy and Structural Biology, Albert Einstein College of Medicine, Bronx, NY 10461 Submitted May 22, 2008; Revised July 14, 2008; Accepted July 15, 2008 Monitoring Editor: Marvin Wickens Exported mRNAs are targeted for translation or can undergo degradation by several decay mechanisms. The 533 degradation machinery localizes to cytoplasmic P bodies (PBs). We followed the dynamic properties of PBs in vivo and investigated the mechanism by which PBs scan the cytoplasm. Using proteins of the decapping machinery, we asked whether PBs actively scan the cytoplasm or whether a diffusion-based mechanism is sufficient. Live-cell imaging showed that PBs were anchored mainly to microtubules. Quantitative single-particle tracking demonstrated that most PBs exhibited spatially confined motion depen- dent on microtubule motion, whereas stationary PB pairs were identified at the centrosome. Some PBs translocated in long-range movements on microtubules. PB mobility was compared with mitochondria, endoplasmic reticulum, peroxisomes, SMN bodies, and stress granules, and diffusion coefficients were calculated. Disruption of the microtubule network caused a significant reduction in PB mobility together with an induction of PB assembly. However, FRAP measurements showed that the dynamic flux of assembled PB components was not affected by such treatments. -

Bardaweel Umn 0130E 11654.Pdf (7.944Mb Application/Pdf)
DEFINING THE CATALYTIC AND KINETIC MECHANISM AND NATURAL FUNCTION OF THE HIGHLY CONSERVED ACYL-AMP HYDROLASE, HINT1 A DISSERTATION SUBMITTED TO THE FACULTY OF THE GRADUATE SCHOOL OF THE UNIVERSITY OF MINNESOTA BY SANAA BARDAWEEL IN PARTIAL FULFILMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTORE OF PHILOSOPHY DR. CARSTON R. WAGNER, ADVISER DECEMBER 2010 © Sanaa Bardaweel, 2010 ACKNOWLEDGEMENTS I would like to gratefully and sincerely thank my thesis adviser, Dr. Carston R. Wagner, for his guidance, understanding and patience. His mentorship was paramount in providing a well rounded experience consistent with my long-term career goals. He encouraged me to not only grow as an experimentalist but also as an instructor and an independent researcher. In every sense, none of this work would have been possible without him. I gratefully acknowledge Dr. Michael Sadowsky for his advice, supervision, and crucial contributions, which made him a backbone of this research and so to this thesis. I am also deeply thankful to Dr. Patrick Hanna, for the valuable advice and discussions that inspired me through out the work. Many thanks must also go to my thesis committee members, Dr. Chengguo Xing and Dr. Howard Towle, for their valuable input and suggestions that improved my dissertation. I would like to make a special reference to Dr. Yusuf Abul-Hajj for his unflinching encouragement and support in various ways. His support and guidance during the very first steps in my PhD have had a remarkable influence on my entire progress in the program. I am grateful to former members of Dr. Wagner’s laboratory, Dr. -
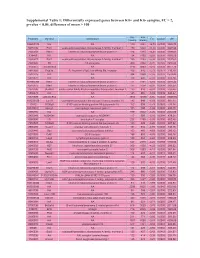
Differentially Expressed Genes Between Kit+ and Kit- Samples, FC
Supplemental Table 1: Differentially expressed genes between Kit+ and Kit- samples, FC > 2, p-value < 0.05, difference of mean > 100 Kit- Kit+ Probeset Symbol Genename FC pvalue diff (mean) (mean) 104280139 NA NA 130 1651 -12,70 0,0000 1520,78 5670239 Ear2 eosinophil-associated, ribonuclease A family, member 2 150 1848 -12,28 0,0000 1697,94 2340358 Ifitm3 interferon induced transmembrane protein 3 126 1315 -10,41 0,0000 1189,02 130465 NA NA 134 1155 -8,60 0,0000 1020,25 2360471 Ear1 eosinophil-associated, ribonuclease A family, member 1 155 1293 -8,34 0,0000 1137,61 7040095 Kit kit oncogene 409 3364 -8,22 0,0145 2955,05 1230347 LOC545854 NA 1216 9893 -8,14 0,0000 8677,76 2810059 Fcgr3a Fc fragment of IgG, low affinity IIIa, receptor 106 816 -7,73 0,0077 710,20 2510725 NA NA 394 2969 -7,53 0,0037 2574,43 5420372 NA NA 128 942 -7,33 0,0000 813,19 101990390 Ifitm2 interferon induced transmembrane protein 2 171 1181 -6,93 0,0000 1010,43 6510075 Ifitm1 interferon induced transmembrane protein 1 152 1054 -6,92 0,0004 901,68 2370286 Slc40a1 solute carrier family 40 (iron-regulated transporter), member 1 133 916 -6,91 0,0000 783,46 5860673 NA NA 143 985 -6,89 0,0239 842,27 6370309 LOC545854 NA 1850 12680 -6,85 0,0000 10829,66 101230129 Ear10 eosinophil-associated, ribonuclease A family, member 10 142 949 -6,68 0,0000 807,11 70112 S100a8 S100 calcium binding protein A8 (calgranulin A) 162 938 -5,79 0,0063 776,16 103780671 Mpeg1 macrophage expressed gene 1 105 598 -5,69 0,0000 493,10 1690184 NA NA 579 3042 -5,26 0,0002 2463,90 2450148 AI324046 expressed -

Life and Death of Mrna Molecules in Entamoeba Histolytica
REVIEW published: 19 June 2018 doi: 10.3389/fcimb.2018.00199 Life and Death of mRNA Molecules in Entamoeba histolytica Jesús Valdés-Flores 1, Itzel López-Rosas 2, César López-Camarillo 3, Esther Ramírez-Moreno 4, Juan D. Ospina-Villa 4† and Laurence A. Marchat 4* 1 Departamento de Bioquímica, CINVESTAV, Ciudad de Mexico, Mexico City, Mexico, 2 CONACyT Research Fellow – Colegio de Postgraduados Campus Campeche, Campeche, Mexico, 3 Posgrado en Ciencias Genómicas, Universidad Autónoma de la Ciudad de México, Ciudad de Mexico, Mexico City, Mexico, 4 Escuela Nacional de Medicina y Homeopatía, Instituto Politécnico Nacional, Ciudad de Mexico, Mexico City, Mexico In eukaryotic cells, the life cycle of mRNA molecules is modulated in response to environmental signals and cell-cell communication in order to support cellular homeostasis. Capping, splicing and polyadenylation in the nucleus lead to the formation of transcripts that are suitable for translation in cytoplasm, until mRNA decay occurs Edited by: in P-bodies. Although pre-mRNA processing and degradation mechanisms have usually Mario Alberto Rodriguez, been studied separately, they occur simultaneously and in a coordinated manner through Centro de Investigación y de Estudios Avanzados del Instituto Politécnico protein-protein interactions, maintaining the integrity of gene expression. In the past few Nacional (CINVESTAV-IPN), Mexico years, the availability of the genome sequence of Entamoeba histolytica, the protozoan Reviewed by: parasite responsible for human amoebiasis, coupled to the development of the so-called Mark R. Macbeth, “omics” technologies provided new opportunities for the study of mRNA processing and Butler University, United States Michael G. Sehorn, turnover in this pathogen.