Identification of the Hub Genes and Molecular Pathways Involved in SIRS Based on Bioinformatics Analysis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pan-KIR2DL NK-Receptor Antibodies and Their Use in Diagnostik and Therapy
(19) TZZ _ T (11) EP 2 287 195 A2 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 23.02.2011 Bulletin 2011/08 C07K 16/28 (2006.01) G01N 33/52 (2006.01) (21) Application number: 10178924.6 (22) Date of filing: 01.07.2005 (84) Designated Contracting States: • Romagne, François AT BE BG CH CY CZ DE DK EE ES FI FR GB GR 13600 La Ciotat (FR) HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI • Wagtmann, Peter, Andreas, Nicolai, Reumert SK TR 2960 Rungsted Kyst (DK) • Svendsen, Ivan (30) Priority: 06.01.2005 DK 200500025 2765 Smorum (DK) 01.07.2004 PCT/DK2004/000470 • Zahn, Stefan 01.07.2004 PCT/IB2004/002464 2750 Ballerup (DK) • Svensson, Anders (62) Document number(s) of the earlier application(s) in 21746 Malmö (DK) accordance with Art. 76 EPC: • Thorolfsson, Matthias 05758642.2 / 1 791 868 2920 Charlottenlund (DK) • Berg Padkaer, Soren (71) Applicants: 3500 Vaerlose (DK) • Novo Nordisk A/S • Kjaergaard, Kristian 2880 Bagsvaerd (DK) 2750 Ballerup (DK) • Innate Pharma • Spee, Pieter 13009 Marseille (FR) 3450 Allerod (DK) • Universita di Genova • Wilken, Michael 16132 Genova (IT) 3390 Hundested (DK) (72) Inventors: (74) Representative: Gallois, Valérie et al • Moretta, Alessandro Cabinet BECKER & ASSOCIES 16133 Genova (IT) 25, rue Louis Le Grand • Della Chiesa, Mariella 75002 Paris (FR) 16132 Genova (IT) • Andre, Pascale Remarks: 13006 Marseille (FR) This application was filed on 23-09-2010 as a • Gauthier, Laurent divisional application to the application mentioned 13008 Marseille (FR) under INID code 62. -

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -

Flow Reagents Single Color Antibodies CD Chart
CD CHART CD N° Alternative Name CD N° Alternative Name CD N° Alternative Name Beckman Coulter Clone Beckman Coulter Clone Beckman Coulter Clone T Cells B Cells Granulocytes NK Cells Macrophages/Monocytes Platelets Erythrocytes Stem Cells Dendritic Cells Endothelial Cells Epithelial Cells T Cells B Cells Granulocytes NK Cells Macrophages/Monocytes Platelets Erythrocytes Stem Cells Dendritic Cells Endothelial Cells Epithelial Cells T Cells B Cells Granulocytes NK Cells Macrophages/Monocytes Platelets Erythrocytes Stem Cells Dendritic Cells Endothelial Cells Epithelial Cells CD1a T6, R4, HTA1 Act p n n p n n S l CD99 MIC2 gene product, E2 p p p CD223 LAG-3 (Lymphocyte activation gene 3) Act n Act p n CD1b R1 Act p n n p n n S CD99R restricted CD99 p p CD224 GGT (γ-glutamyl transferase) p p p p p p CD1c R7, M241 Act S n n p n n S l CD100 SEMA4D (semaphorin 4D) p Low p p p n n CD225 Leu13, interferon induced transmembrane protein 1 (IFITM1). p p p p p CD1d R3 Act S n n Low n n S Intest CD101 V7, P126 Act n p n p n n p CD226 DNAM-1, PTA-1 Act n Act Act Act n p n CD1e R2 n n n n S CD102 ICAM-2 (intercellular adhesion molecule-2) p p n p Folli p CD227 MUC1, mucin 1, episialin, PUM, PEM, EMA, DF3, H23 Act p CD2 T11; Tp50; sheep red blood cell (SRBC) receptor; LFA-2 p S n p n n l CD103 HML-1 (human mucosal lymphocytes antigen 1), integrin aE chain S n n n n n n n l CD228 Melanotransferrin (MT), p97 p p CD3 T3, CD3 complex p n n n n n n n n n l CD104 integrin b4 chain; TSP-1180 n n n n n n n p p CD229 Ly9, T-lymphocyte surface antigen p p n p n -

Supplementary Material DNA Methylation in Inflammatory Pathways Modifies the Association Between BMI and Adult-Onset Non- Atopic
Supplementary Material DNA Methylation in Inflammatory Pathways Modifies the Association between BMI and Adult-Onset Non- Atopic Asthma Ayoung Jeong 1,2, Medea Imboden 1,2, Akram Ghantous 3, Alexei Novoloaca 3, Anne-Elie Carsin 4,5,6, Manolis Kogevinas 4,5,6, Christian Schindler 1,2, Gianfranco Lovison 7, Zdenko Herceg 3, Cyrille Cuenin 3, Roel Vermeulen 8, Deborah Jarvis 9, André F. S. Amaral 9, Florian Kronenberg 10, Paolo Vineis 11,12 and Nicole Probst-Hensch 1,2,* 1 Swiss Tropical and Public Health Institute, 4051 Basel, Switzerland; [email protected] (A.J.); [email protected] (M.I.); [email protected] (C.S.) 2 Department of Public Health, University of Basel, 4001 Basel, Switzerland 3 International Agency for Research on Cancer, 69372 Lyon, France; [email protected] (A.G.); [email protected] (A.N.); [email protected] (Z.H.); [email protected] (C.C.) 4 ISGlobal, Barcelona Institute for Global Health, 08003 Barcelona, Spain; [email protected] (A.-E.C.); [email protected] (M.K.) 5 Universitat Pompeu Fabra (UPF), 08002 Barcelona, Spain 6 CIBER Epidemiología y Salud Pública (CIBERESP), 08005 Barcelona, Spain 7 Department of Economics, Business and Statistics, University of Palermo, 90128 Palermo, Italy; [email protected] 8 Environmental Epidemiology Division, Utrecht University, Institute for Risk Assessment Sciences, 3584CM Utrecht, Netherlands; [email protected] 9 Population Health and Occupational Disease, National Heart and Lung Institute, Imperial College, SW3 6LR London, UK; [email protected] (D.J.); [email protected] (A.F.S.A.) 10 Division of Genetic Epidemiology, Medical University of Innsbruck, 6020 Innsbruck, Austria; [email protected] 11 MRC-PHE Centre for Environment and Health, School of Public Health, Imperial College London, W2 1PG London, UK; [email protected] 12 Italian Institute for Genomic Medicine (IIGM), 10126 Turin, Italy * Correspondence: [email protected]; Tel.: +41-61-284-8378 Int. -

High-Dimensional Analysis Reveals a Distinct Population of Adaptive-Like Tissue
bioRxiv preprint doi: https://doi.org/10.1101/2019.12.20.883785; this version posted December 20, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 High-dimensional analysis reveals a distinct population of adaptive-like tissue- 2 resident NK cells in human lung 3 4 Nicole Marquardt1*, Marlena Scharenberg1, Jeffrey E. Mold2, Joanna Hård2, Eliisa 5 Kekäläinen3,4, Marcus Buggert1, Son Nguyen5,6, Jennifer N. Wilson1, Mamdoh Al- 6 Ameri7, Hans-Gustaf Ljunggren1, Jakob Michaëlsson1 7 8 Running title: Adaptive-like human lung tissue-resident NK cells 9 10 Affiliations: 11 1Center for Infectious Medicine, Department of Medicine, Karolinska Institutet, 12 Stockholm, Sweden 13 2Department of Cell and Molecular Biology, Karolinska Institutet, Stockholm, Sweden 14 3Translational Immunology Research Program & Department of Bacteriology and 15 Immunology, University of Helsinki, Finland 16 4HUSLAB, Division of Clinical Microbiology, Helsinki University Hospital, Helsinki, 17 Finland, 18 5Department of Microbiology, Perelman School of Medicine, University of 19 Pennsylvania, Philadelphia, PA, USA 20 6Institute for Immunology, Perelman School of Medicine, University of Pennsylvania, 21 Philadelphia, PA, USA 22 7Thoracic Surgery, Department of Molecular Medicine and Surgery, Karolinska 23 Institutet, Karolinska University Hospital, Stockholm, Sweden 24 1 bioRxiv preprint doi: https://doi.org/10.1101/2019.12.20.883785; this version posted December 20, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Systems Analysis of Protective Immune Responses to RTS,S Malaria Vaccination in Humans
Systems analysis of protective immune responses to RTS,S malaria vaccination in humans Dmitri Kazmina,1, Helder I. Nakayab,1, Eva K. Leec, Matthew J. Johnsond, Robbert van der Moste, Robert A. van den Bergf, W. Ripley Ballouf, Erik Jongerte, Ulrike Wille-Reeceg, Christian Ockenhouseg, Alan Aderemh, Daniel E. Zakh, Jerald Sadoffi, Jenny Hendriksi, Jens Wrammerta, Rafi Ahmeda,2, and Bali Pulendrana,j,2 aEmory Vaccine Center, Emory University, Atlanta, GA 30329; bSchool of Pharmaceutical Sciences, University of Sao Paulo, São Paulo 05508, Brazil; cSchool of Industrial and Systems Engineering, Georgia Institute of Technology, Atlanta, GA 30332; dCenter for Genome Engineering, University of Minnesota, Minneapolis, MN 55108; eGSK Vaccines, Rixensart 1330, Belgium; fGSK Vaccines, Rockville, MD 20850; gProgram for Appropriate Technology in Health-Malaria Vaccine Initiative, Washington, DC 20001; hCenter for Infectious Disease Research, Seattle, WA 98109; iCrucell, Leiden 2333, The Netherlands; and jDepartment of Pathology, Emory University School of Medicine, Atlanta, GA 30322 Contributed by Rafi Ahmed, January 4, 2017 (sent for review December 19, 2016; reviewed by Elias Haddad and Robert Seder) RTS,S is an advanced malaria vaccine candidate and confers trials conducted in malaria endemic areas in Africa proved the significant protection against Plasmodium falciparum infection in vaccine to be partially protective in adults (9), children (10, 11), humans. Little is known about the molecular mechanisms driving and infants (12, 13). These results were further confirmed in a vaccine immunity. Here, we applied a systems biology approach phase III trial in sub-Saharan Africa (14–17) in which 55.8% to study immune responses in subjects receiving three consecutive efficacy against clinical malaria was observed over the first 12 mo immunizations with RTS,S (RRR), or in those receiving two immuni- of follow-up in children of 5–17 mo (14). -

Natural Killer Cell Lymphoma Shares Strikingly Similar Molecular Features
Leukemia (2011) 25, 348–358 & 2011 Macmillan Publishers Limited All rights reserved 0887-6924/11 www.nature.com/leu ORIGINAL ARTICLE Natural killer cell lymphoma shares strikingly similar molecular features with a group of non-hepatosplenic cd T-cell lymphoma and is highly sensitive to a novel aurora kinase A inhibitor in vitro J Iqbal1, DD Weisenburger1, A Chowdhury2, MY Tsai2, G Srivastava3, TC Greiner1, C Kucuk1, K Deffenbacher1, J Vose4, L Smith5, WY Au3, S Nakamura6, M Seto6, J Delabie7, F Berger8, F Loong3, Y-H Ko9, I Sng10, X Liu11, TP Loughran11, J Armitage4 and WC Chan1, for the International Peripheral T-cell Lymphoma Project 1Department of Pathology and Microbiology, University of Nebraska Medical Center, Omaha, NE, USA; 2Eppley Institute for Research in Cancer and Allied Diseases, University of Nebraska Medical Center, Omaha, NE, USA; 3Departments of Pathology and Medicine, University of Hong Kong, Queen Mary Hospital, Hong Kong, China; 4Division of Hematology and Oncology, Department of Internal Medicine, University of Nebraska Medical Center, Omaha, NE, USA; 5College of Public Health, University of Nebraska Medical Center, Omaha, NE, USA; 6Departments of Pathology and Cancer Genetics, Aichi Cancer Center Research Institute, Nagoya University, Nagoya, Japan; 7Department of Pathology, University of Oslo, Norwegian Radium Hospital, Oslo, Norway; 8Department of Pathology, Centre Hospitalier Lyon-Sud, Lyon, France; 9Department of Pathology, Samsung Medical Center, Sungkyunkwan University, Seoul, Korea; 10Department of Pathology, Singapore General Hospital, Singapore and 11Penn State Hershey Cancer Institute, Pennsylvania State University College of Medicine, Hershey, PA, USA Natural killer (NK) cell lymphomas/leukemias are rare neo- Introduction plasms with an aggressive clinical behavior. -

Molecular Signatures Differentiate Immune States in Type 1 Diabetes Families
Page 1 of 65 Diabetes Molecular signatures differentiate immune states in Type 1 diabetes families Yi-Guang Chen1, Susanne M. Cabrera1, Shuang Jia1, Mary L. Kaldunski1, Joanna Kramer1, Sami Cheong2, Rhonda Geoffrey1, Mark F. Roethle1, Jeffrey E. Woodliff3, Carla J. Greenbaum4, Xujing Wang5, and Martin J. Hessner1 1The Max McGee National Research Center for Juvenile Diabetes, Children's Research Institute of Children's Hospital of Wisconsin, and Department of Pediatrics at the Medical College of Wisconsin Milwaukee, WI 53226, USA. 2The Department of Mathematical Sciences, University of Wisconsin-Milwaukee, Milwaukee, WI 53211, USA. 3Flow Cytometry & Cell Separation Facility, Bindley Bioscience Center, Purdue University, West Lafayette, IN 47907, USA. 4Diabetes Research Program, Benaroya Research Institute, Seattle, WA, 98101, USA. 5Systems Biology Center, the National Heart, Lung, and Blood Institute, the National Institutes of Health, Bethesda, MD 20824, USA. Corresponding author: Martin J. Hessner, Ph.D., The Department of Pediatrics, The Medical College of Wisconsin, Milwaukee, WI 53226, USA Tel: 011-1-414-955-4496; Fax: 011-1-414-955-6663; E-mail: [email protected]. Running title: Innate Inflammation in T1D Families Word count: 3999 Number of Tables: 1 Number of Figures: 7 1 For Peer Review Only Diabetes Publish Ahead of Print, published online April 23, 2014 Diabetes Page 2 of 65 ABSTRACT Mechanisms associated with Type 1 diabetes (T1D) development remain incompletely defined. Employing a sensitive array-based bioassay where patient plasma is used to induce transcriptional responses in healthy leukocytes, we previously reported disease-specific, partially IL-1 dependent, signatures associated with pre and recent onset (RO) T1D relative to unrelated healthy controls (uHC). -

Receptors of CD4 T Cells Expressing Killer Ig-Like Phenotypic And
The Journal of Immunology Phenotypic and Functional Characterization of CD4 T Cells Expressing Killer Ig-Like Receptors1 Jeroen van Bergen,2* Allan Thompson,* Arno van der Slik,* Tom H. M. Ottenhoff,* Jacobijn Gussekloo,† and Frits Koning* Killer Ig-like receptors (KIR) are commonly found on human NK cells, ␥␦ T cells, and CD8 T cells. Although KIR؉ CD4 T cells are found in certain patients, their prevalence in healthy donors is controversial. We now provide definitive proof that such cells are present in most individuals, and report on their frequency, surface phenotype, cytokine profile, and Ag specificity. The number of KIR؉ CD4 T cells detected in peripheral blood increased with age. In contrast with regular KIR؊ CD4 T cells, the majority ,of KIR؉ CD4 T cells lacked surface expression of CD27, CD28, CCR4, and CCR7, but did express CD57 and 2B4. In addition KIR were detected on approximately one-tenth of CD28؊ and CD57؉ memory CD4 T cells. In line with the absence of the Th2 ,marker CCR4, the KIR؉ CD4 cells produced mainly IFN-␥ and little IL-4, IL-10, or IL-17 upon TCR triggering. Furthermore the KIR؉ population contained cells that responded to recall Ags in an HLA class II-restricted fashion. Together, our data indicate that KIR-expressing CD4 T cells are predominantly HLA class II-restricted effector memory Th1 cells, and that a significant, previously unrecognized fraction of effector memory Th1 cells expresses KIR. The Journal of Immunology, 2004, 173: 6719–6726. atural killer cells use a wide variety of surface receptors KIR expression by CD4 T cells is scarce. -

Expansions of Adaptive-Like NK Cells with a Tissue-Resident Phenotype in Human Lung and Blood
Expansions of adaptive-like NK cells with a tissue-resident phenotype in human lung and blood Demi Brownliea,1, Marlena Scharenberga,1, Jeff E. Moldb, Joanna Hårdb, Eliisa Kekäläinenc,d,e, Marcus Buggerta, Son Nguyenf,g, Jennifer N. Wilsona, Mamdoh Al-Amerih, Hans-Gustaf Ljunggrena, Nicole Marquardta,2,3, and Jakob Michaëlssona,2 aCenter for Infectious Medicine, Department of Medicine Huddinge, Karolinska Institutet, 14152 Stockholm, Sweden; bDepartment of Cell and Molecular Biology, Karolinska Institutet, 171 77 Stockholm, Sweden; cTranslational Immunology Research Program, University of Helsinki, 00014 Helsinki, Finland; dDepartment of Bacteriology and Immunology, University of Helsinki, 00014 Helsinki, Finland; eHelsinki University Central Hospital Laboratory, Division of Clinical Microbiology, Helsinki University Hospital, 00290 Helsinki, Finland; fDepartment of Microbiology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104; gInstitute for Immunology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104; and hThoracic Surgery, Department of Molecular Medicine and Surgery, Karolinska University Hospital, Karolinska Institutet, 171 76 Stockholm, Sweden Edited by Marco Colonna, Washington University in St. Louis School of Medicine, St. Louis, MO, and approved January 27, 2021 (received for review August 18, 2020) Human adaptive-like “memory” CD56dimCD16+ natural killer (NK) We and others recently identified a subset of tissue-resident − cells in peripheral blood from cytomegalovirus-seropositive indi- CD49a+CD56brightCD16 NK cells in the human lung (14, 15). viduals have been extensively investigated in recent years and are The human lung is a frequent site of infection with viruses such currently explored as a treatment strategy for hematological can- as influenza virus and HCMV, as well as a reservoir for latent cers. -
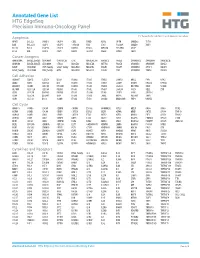
Annotated Gene List HTG Edgeseq Precision Immuno-Oncology Panel
Annotated Gene List HTG EdgeSeq Precision Immuno-Oncology Panel For Research Use Only. Not for use in diagnostic procedures. Apoptosis APAF1 BCL2L1 CARD11 CASP4 CD5L FADD KSR2 OPTN SAMD12 TCF19 BAX BCL2L11 CASP1 CASP5 CORO1A FAS LRG1 PLA2G6 SAMD9 XAF1 BCL10 BCL6 CASP10 CASP8 DAPK2 FASLG MECOM PYCARD SPOP BCL2 BID CASP3 CAV1 DAPL1 GLIPR1 MELK RIPK2 TBK1 Cancer Antigens ANKRD30A BAGE2_BAGE3 CEACAM6 CTAG1A_1B LIPE MAGEA3_A6 MAGEC2 PAGE3 SPANXACD SPANXN4 XAGE1B_1E ARMCX6 BAGE4_BAGE5 CEACAM8 CTAG2 MAGEA1 MAGEA4 MTFR2 PAGE4 SPANXB1 SPANXN5 XAGE2 BAGE CEACAM1 CT45_family GAGE_family MAGEA10 MAGEB2 PAGE1 PAGE5 SPANXN1 SYCP1 XAGE3 BAGE_family CEACAM5 CT47_family HPN MAGEA12 MAGEC1 PAGE2 PBK SPANXN3 TEX14 XAGE5 Cell Adhesion ADAM17 CDH15 CLEC5A DSG3 ICAM2 ITGA5 ITGB2 LAMC3 MBL2 PVR UPK2 ADD2 CDH5 CLEC6A DST ICAM3 ITGA6 ITGB3 LAMP1 MTDH RRAS2 UPK3A ADGRE5 CLDN3 CLEC7A EPCAM ICAM4 ITGAE ITGB4 LGALS1 NECTIN2 SELE VCAM1 ALCAM CLEC12A CLEC9A FBLN1 ITGA1 ITGAL ITGB7 LGALS3 OCLN SELL ZYX CD63 CLEC2B DIAPH3 FXYD5 ITGA2 ITGAM ITLN2 LYVE1 OLR1 SELPLG CD99 CLEC4A DLGAP5 IBSP ITGA3 ITGAX JAML M6PR PECAM1 THY1 CDH1 CLEC4C DSC3 ICAM1 ITGA4 ITGB1 L1CAM MADCAM1 PKP1 UNC5D Cell Cycle ANAPC1 CCND3 CDCA5 CENPH CNNM1 ESCO2 HORMAD2 KIF2C MELK ORC6 SKA3 TPX2 ASPM CCNE1 CDCA8 CENPI CNTLN ESPL1 IKZF1 KIF4A MND1 PATZ1 SP100 TRIP13 AURKA CCNE2 CDK1 CENPL CNTLN ETS1 IKZF2 KIF5C MYBL2 PIF1 SP110 TROAP AURKB CCNF CDK4 CENPU DBF4 ETS2 IKZF3 KIFC1 NCAPG PIMREG SPC24 TUBB BEX1 CDC20 CDK6 CENPW E2F2 EZH2 IKZF4 KNL1 NCAPG2 PKMYT1 SPC25 ZWILCH BEX2 CDC25A CDKN1A CEP250 E2F7 GADD45GIP1 -

Review Genetic and Functional Relationships Between MHC and NK Receptor Genes
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Immunity, Vol. 15, 363–374, September, 2001, Copyright 2001 by Cell Press Genetic and Functional Relationships Review between MHC and NK Receptor Genes John Trowsdale1 the human genome, notably at the central class III re- kbp. It 12ف Immunology Division gion, where there is over one gene per Pathology Department encodes the most polymorphic human proteins known University of Cambridge to date, the class I and class II molecules. Of the ex- Tennis Court Road pressed loci in the MHC, roughly 40% are associated Cambridge CB2 1QP with the immune system. Some of the main immune United Kingdom system genes are shown in Figure 2. They include the classical class I, HLA-A, -B, and -C, nonclassical HLA-E, -F, and -G, as well as “postmodern” MICA and HLA class I and NK receptors are encoded within MICB genes (MHC class I chain-related genes). There dense clusters of immune loci. The MHC, at 6p21.3, are also nonexpressed pseudogenes representing all and the complex containing the KIR loci, at 19q13.4, three groups of sequence. The products of classical polymorphic class I genes, HLA-A, B, and C, interact both feature variation in the number of genes, as well with T cell receptor (TCR) molecules as well as with the as sequence polymorphism. In addition to T cell recep- products of the killer immunoglobulin-like receptor (KIR) tors, several variable class I-related molecules interact genes expressed on natural killer cells and some T cells.