Minimally Invasive Prenatal Diagnosis of Inherited Disorders Employing Trophoblastic Cells Shed Into the Endocervical Canal
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Central Intelligence Agency (CIA) Freedom of Information Act (FOIA) Case Log October 2000 - April 2002
Description of document: Central Intelligence Agency (CIA) Freedom of Information Act (FOIA) Case Log October 2000 - April 2002 Requested date: 2002 Release date: 2003 Posted date: 08-February-2021 Source of document: Information and Privacy Coordinator Central Intelligence Agency Washington, DC 20505 Fax: 703-613-3007 Filing a FOIA Records Request Online The governmentattic.org web site (“the site”) is a First Amendment free speech web site and is noncommercial and free to the public. The site and materials made available on the site, such as this file, are for reference only. The governmentattic.org web site and its principals have made every effort to make this information as complete and as accurate as possible, however, there may be mistakes and omissions, both typographical and in content. The governmentattic.org web site and its principals shall have neither liability nor responsibility to any person or entity with respect to any loss or damage caused, or alleged to have been caused, directly or indirectly, by the information provided on the governmentattic.org web site or in this file. The public records published on the site were obtained from government agencies using proper legal channels. Each document is identified as to the source. Any concerns about the contents of the site should be directed to the agency originating the document in question. GovernmentAttic.org is not responsible for the contents of documents published on the website. 1 O ct 2000_30 April 2002 Creation Date Requester Last Name Case Subject 36802.28679 STRANEY TECHNOLOGICAL GROWTH OF INDIA; HONG KONG; CHINA AND WTO 36802.2992 CRAWFORD EIGHT DIFFERENT REQUESTS FOR REPORTS REGARDING CIA EMPLOYEES OR AGENTS 36802.43927 MONTAN EDWARD GRADY PARTIN 36802.44378 TAVAKOLI-NOURI STEPHEN FLACK GUNTHER 36810.54721 BISHOP SCIENCE OF IDENTITY FOUNDATION 36810.55028 KHEMANEY TI LEAF PRODUCTIONS, LTD. -
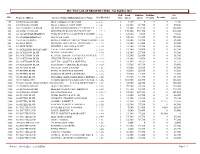
2021 Assessment Roll
INC. VILLAGE OF BRIGHTWATERS - Tax Roll For 2021 Roll Land Building Building Net ID # Property Address Owner's Name/Additional Owner's Name Sect/Block/Lot Type Assess Assess Permits Exempts Assess 100 200 HOWELLS ROAD MJ & JJ REALTY CORP CORP 1- 1- 1- 1 3,500 0 0 0 3,500 200 200 HOWELLS ROAD MK & JJ REALTY CORP CORP 1- 1- 2- 1 12,300 68,300 0 0 80,600 300 180-196 HOWELLS ROAD NK ENTERPRISES/WESLEY Y CHUNG, CP 1- 1- 3- 1 81,100 131,100 0 0 212,200 400 225 HOWELLS ROAD BRIGHTWATERS RACQUET& SPA INC 1- 1- 7-.1 1 170,300 459,700 0 0 630,000 500 969 NO SUNRISE HIGHWAY ZERIS REALTY LLC/C/O PETER PAN DINE 1- 1- 8- 1 64,800 9,600 0 0 74,400 600 985 SUNRISE HIGHWAY 1668 N.Y.A. CORP 1- 1- 9- 1 54,900 51,000 0 0 105,900 700 105 SENECA DRIVE BOWLES RODNEY/TRICIA C DALEY-BOWL1- 1- 12- 1 11,300 33,700 0 0 45,000 800 556 PINE DRIVE WOHNING, LIFE ESTATE GUNTHER H. E 1- 1- 13- 1 11,300 21,000 0 0 32,300 900 552 PINE DRIVE MENNELLA ROLAND & JANET 1- 1- 14- 1 11,300 42,700 0 0 54,000 1000 555 ACKERSON BOULEVARD CAVALLUZZI GERALDINE 1- 1- 15- 1 11,300 34,900 0 0 46,200 1100 563 ACKERSON BLVD DEVINE ROSEMARIE 1- 1- 16- 1 11,300 27,900 0 0 39,200 1200 565 ACKERSON BLVD KEATING JOANNE M/CRAIG KEATING & T1- 1- 17- 1 11,300 26,800 0 0 38,100 1300 562 ACKERSON BLVD CHRISTIE THOMAS & ROBYN T 1- 1- 18- 1 11,300 33,650 0 0 44,950 1400 558 ACKERSON BLVD COLLURA SCOTT N & NOELLE G 1- 1- 19- 1 11,300 30,255 0 0 41,555 1500 554 ACKERSON BLVD SOLUTIONS CARDINAL BUSINESS 1- 1- 20- 1 11,300 42,150 0 0 53,450 1600 551 PETERS BLVD COLBERT JOHN & KATRIN 1- 1- 21- 1 10,000 -

NOSTALGIA, EMOTIONALITY, and ETHNO-REGIONALISM in PONTIC PARAKATHI SINGING by IOANNIS TSEKOURAS DISSERTATION Submitted in Parti
NOSTALGIA, EMOTIONALITY, AND ETHNO-REGIONALISM IN PONTIC PARAKATHI SINGING BY IOANNIS TSEKOURAS DISSERTATION Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Musicology in the Graduate College of the University of Illinois at Urbana-Champaign, 2016 Urbana, Illinois Doctoral Committee: Associate Professor Donna A. Buchanan, Chair Professor Emeritus Thomas Turino Professor Gabriel Solis Professor Maria Todorova ABSTRACT This dissertation explores the multilayered connections between music, emotionality, social and cultural belonging, collective memory, and identity discourse. The ethnographic case study for the examination of all these relations and aspects is the Pontic muhabeti or parakathi. Parakathi refers to a practice of socialization and music making that is designated insider Pontic Greek. It concerns primarily Pontic Greeks or Pontians, the descendants of the 1922 refugees from Black Sea Turkey (Gr. Pontos), and their identity discourse of ethno-regionalism. Parakathi references nightlong sessions of friendly socialization, social drinking, and dialogical participatory singing that take place informally in coffee houses, taverns, and households. Parakathi performances are reputed for their strong Pontic aesthetics, traditional character, rich and aesthetically refined repertoire, and intense emotionality. Singing in parakathi performances emerges spontaneously from verbal socialization and emotional saturation. Singing is described as a confessional expression of deeply personal feelings -

Geochemical Database of Feed Coal and Coal Combustion Products (Ccps) from Five Power Plants in the United States
Geochemical Database of Feed Coal and Coal Combustion Products (CCPs) from Five Power Plants in the United States Selected Coal Utilization References By Kelly L. Conrad and Ronald H. Affolter Pamphlet to accompany Data Series 635 U.S. Department of the Interior U.S. Geological Survey U.S. Department of the Interior KEN SALAZAR, Secretary U.S. Geological Survey Marcia K. McNutt, Director U.S. Geological Survey, Reston, Virginia: 2011 About USGS Products For product and ordering information: World Wide Web: http://www.usgs.gov/pubprod Telephone: 1-888-ASK-USGS For more information on the USGS—the Federal source for science about the Earth, its natural and living resources, natural hazards, and the environment: World Wide Web: http://www.usgs.gov Telephone: 1-888-ASK-USGS About this Product Publishing support provided by Denver Science Publishing Network For more information concerning this publication, contact: Center Director, USGS Central Energy Resources Science Center Box 25046, Mail stop 939 Denver, CO 80225 (303) 236-7775 or visit the Central Energy Resources Science Center Web site at: http://energy.cr.usgs.gov Suggested citation: Affolter, R.H., Groves, Steve, Betterton, W.J., Benzel, William, Conrad, K.L., Swanson, S.M., Ruppert L.F., Clough J.G., Belkin, H.E., Kolker, Allan, and Hower, J.C., 2011, Geochemical database of feed coal and coal combustion products (CCPs) from five power plants in the United States: U.S. Geological Survey Data Series 635, pamphlet, 19 p. Any use of trade, product, or firm names is for descriptive purposes only and does not imply endorsement by the U.S. -

2021 Village of Greendale Preliminary Value Listing - Street Order
2021 VILLAGE OF GREENDALE PRELIMINARY VALUE LISTING - STREET ORDER PARCEL OWNER_NAME_1 CLASS STREET_# STREET_NAME ACRE LAND BLDG TOTAL 6650142000 GREENDALE, VILLAGE OF EXM 51ST ST S 0.84 0 0 0 6940229000 GREENDALE, VILLAGE OF EXM 51ST ST S 0.62 0 0 0 6480013002 RYAN REVOCABLE TRUST RES 5311 51ST ST S 0.18 63,800 174,300 238,100 6480013003 PALECHEK, MICHAEL P RES 5321 51ST ST S 0.18 63,800 168,400 232,200 6480001002 GLISCH, MARK K & LORI J RES 5339 51ST ST S 0.21 51,100 145,700 196,800 6480002000 HAWKINS, THERESE RES 5369 51ST ST S 0.18 55,600 134,000 189,600 6480044000 BALLARD, CORY E RES 5379 51ST ST S 0.27 51,800 188,700 240,500 6480003000 ARNOLD, BETTY ANN D RES 5389 51ST ST S 0.18 55,600 148,900 204,500 6480004000 PHILLIPS, CHRISTOPHER J RES 5401 51ST ST S 0.19 56,500 123,200 179,700 6480045000 HERING, MICHAEL J & MONIKA A RES 5421 51ST ST S 0.22 51,300 149,400 200,700 6480046000 GAMROTH, MARK RES 5431 51ST ST S 0.22 51,300 153,400 204,700 6480047000 CALABRIA, JAMES R RES 5441 51ST ST S 0.22 51,300 156,500 207,800 6480048000 BRESINA, LORY A RES 5451 51ST ST S 0.25 51,700 148,000 199,700 6659998001 GREENDALE BAPTIST CHURCH EXM 5651 51ST ST S 2.90 0 0 0 6939998002 GREENDALE SCHOOL DISTRICT EXM 5900 51ST ST S 12.44 0 0 0 1 OF 313 2021 VILLAGE OF GREENDALE PRELIMINARY VALUE LISTING - STREET ORDER PARCEL OWNER_NAME_1 CLASS STREET_# STREET_NAME ACRE LAND BLDG TOTAL 6949998006 ST STEPHEN THE MARTYR, LUTHERAN CHURCH EXM 6101 51ST ST S 3.00 0 0 0 6930202001 COLLEGE SQUARE LLC COM 6210 51ST ST S 4.49 600,000 2,535,400 3,135,400 6480016006 GREENDALE, -

2021 Village of Greendale Preliminary Value Listing - Parcel Order
2021 VILLAGE OF GREENDALE PRELIMINARY VALUE LISTING - PARCEL ORDER PARCEL OWNER_NAME_1 CLASS STREET_# STREET_NAME ACRE LAND BLDG TOTAL 6150001000 DAUER, TIMOTHY & ANDREA RES 8835 FOREST HOME AVE W 0.57 72,400 115,800 188,200 6150002000 BAHLING, JAMIE RES 8855 FOREST HOME AVE W 0.48 70,500 68,800 139,300 6150003000 VAN HEESCH, ANDREW RES 8865 FOREST HOME AVE W 0.47 70,100 95,800 165,900 6150004000 BAILEY, ADAM T RES 8875 FOREST HOME AVE W 0.46 69,900 149,200 219,100 6150005000 VEKIC, MIRKO & TANJA RES 8895 FOREST HOME AVE W 0.90 77,100 73,000 150,100 6150006000 PETHKE, KELLY RES 8950 EDGERTON AVE W 0.51 71,000 173,600 244,600 6150007000 BYKOWSKI, JOSEPH L & RENEE RES 8930 EDGERTON AVE W 0.56 72,200 140,200 212,400 6150008000 NOWAK, DAWN E RES 8816 EDGERTON AVE W 0.52 71,300 141,500 212,800 6150009000 HANSEN, CARLTON J RES 8935 MIDLAND DR W 0.57 76,100 168,600 244,700 6150010000 MICHAEL & CHRISTINE KING REVOCABLE TRUST RES 8895 MIDLAND DR W 0.47 72,000 128,500 200,500 6150011000 HANN TRUSTEE 11/1/2012, WILLIAM D RES 8875 MIDLAND DR W 0.47 72,000 124,900 196,900 6150012000 PIATEK, EDMUND R & WIFE RES 8865 MIDLAND DR W 0.65 79,800 152,500 232,300 6150013000 HANCOCK, E ALLEN & JANIS M RES 8855 MIDLAND DR W 0.48 72,300 156,600 228,900 6150014000 BELOT, COURTNEY B RES 8721 MIDLAND DR W 0.52 74,000 105,400 179,400 6150015000 WHALEN, MICHAEL W & SHARYL A RES 8705 MIDLAND DR W 0.62 78,400 161,500 239,900 1 OF 313 2021 VILLAGE OF GREENDALE PRELIMINARY VALUE LISTING - PARCEL ORDER PARCEL OWNER_NAME_1 CLASS STREET_# STREET_NAME ACRE LAND BLDG TOTAL -

ASIC 06A/06, Monday, 20 February 2006 Published by ASIC ASIC Gazette
Commonwealth of Australia Commonwealth of Australia Gazette No. ASIC 06A/06, Monday, 20 February 2006 Published by ASIC ASIC Gazette Contents Unclaimed Consideration for Compulsory Acquisition - S668A Corporations Act RIGHTS OF REVIEW Persons affected by certain decisions made by ASIC under the Corporations Act and the other legislation administered by ASIC may have rights of review. ASIC has published Practice Note 57 [PN57] Notification of rights of review and Information Sheet [INFO 1100] ASIC decisions – your rights to assist you to determine whether you have a right of review. You can obtain a copy of these documents from the ASIC Digest, the ASIC website at www.asic.gov.au or from the Administrative Law Co-ordinator in the ASIC office with which you have been dealing. ISSN 1445-6060 (Online version) Available from www.asic.gov.au ISSN 1445-6079 (CD-ROM version) Email [email protected] © Commonwealth of Australia, 2006 This work is copyright. Apart from any use permitted under the Copyright Act 1968, all rights are reserved. Requests for authorisation to reproduce, publish or communicate this work should be made to: Gazette Publisher, Australian Securities and Investment Commission, GPO Box 9827, Melbourne Vic 3001 Commonwealth of Australia Gazette ASIC Gazette ASIC 06A/06, Monday, 20 February 2006 Unclaimed Consideration for Compulsory Acquisition Page 2 of 411 Unclaimed Consideration for Compulsory Acquisition - S668A Corporations Act Copies of records of unclaimed consideration in respect of securities, of the following -

GOVERNMENT of the DISTRICT of COLUMBIA Department of Health Care Finance -*** Office of the Senior Deputy/State Medicaid Director Transmittal# 17-28
GOVERNMENT OF THE DISTRICT OF COLUMBIA Department of Health Care Finance -*** Office of the Senior Deputy/State Medicaid Director Transmittal# 17-28 To: District of Columbia Medicaid Managed Care Organizations (MCO) From: Claudia Schlosberg, Esq. ~S Senior Deputy Director and State Medicaid Director Date: November 7, 2017 Subject: Mandatory Medicaid Managed Care Network Provider Enrollment in D.C. Medicaid The 21st Century Cures Act requires all Medicaid Managed Care network providers to enroll in State Medicaid programs by January 1, 2018. Section C.5.21.1.15 of your Managed Care contract with the Department of Health Care Finance (DHCF) also requires network provider enrollment with the D.C. Medicaid program. Appendix A of this transmittal lists the Medicaid Managed Care network provider types that must enroll with the D.C. Medicaid program. Appendix B of this transmittal contains a list of all providers who are currently enrolled in the D.C. Medicaid program. By November 9, 2017, please utilize this list to identify the specific providers that need to enroll in the D.C. Medicaid program. Please send this list by e-mail to Lawrence Williams (DHCF) at [email protected]. Appendix C of this transmittal contains a sample MCO Outreach letter. Please distribute the letter electronically to the network providers you identified by November 10, 2017. The outreach letter informs applicable network providers that their D.C. Medicaid applications are due by November 30,2017. The outreach letter also explains that only one D.C. Medicaid application is required even if they receive the same outreach letter from multiple Managed Care plans. -

Index to 1943 Obituaries in the Canton Repository
Index to 1943 Obituaries in the Canton Repository Published by the Stark County District Library, 2005 Canton, OH Page 1 Index to 1943 Obituaries in the Canton Repository The following is an alphabetical listing, by the deceased’s surname, of obituaries that appeared in the Canton, Ohio newspaper, The Repository. It was compiled by the library staff during the course of the year in the hope that it would be a useful tool to genealogy, as well as other researchers. To use this index, simply locate the name of the person whose obituary you wish to find. The entries will appear as follows: Miller William A. (Estella M.) Mrs. 1940 Jan. 13 30 The first column is the name of the deceased. The second gives the date on which the item appeared. And the third tells the page number containing the obituary. You may find one name with multiple listings showing different dates and locations in the paper. This indicates consecutive listings for that individual. I encourage you to view each for additional information. Page 2 Surname Given Name Maiden Title Year Mth. Day Pg. Abbuhl Bertha Mrs. 1943 Mar. 3 4 Abraham George 1943 Sept. 29 5 Abrams William H. 1943 Oct. 14 10 Ackerman Carl B. Dr. 1943 Jun. 9 11 Ackerman Ephraim (Nellie) Blake Mrs. 1943 Aug. 8 9 Ackerman Harry 1943 Aug. 26 12 Ackerman Nellie F. Mrs. 1943 Dec. 4 2 Ackerman Nellie F. Mrs. 1943 Dec. 5 17 Adams Eli J. 1943 Jun. 11 11 Adams J. Chester 1943 Feb. 13 8 Adams Kenneth M. -
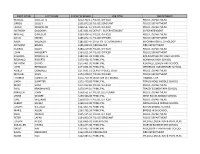
First Name Last Name Salary & Wages Job Title Department
FIRST NAME LAST NAME SALARY & WAGES JOB TITLE DEPARTMENT RUSSELL OUELLETTE $212,702.11 POLICE OFFICER POLICE DEPARTMENT GREGG SCULLY $205,873.26 POLICE SERGEANT POLICE DEPARTMENT JAVIER MOGOLLON $198,547.61 POLICE OFFICER POLICE DEPARTMENT ANTHONY DADDONA $197,691.02 DEPUTY SUPERINTENDENT SUPERINTENDENT MICHAEL DIMEGLIO $190,024.41 POLICE OFFICER POLICE DEPARTMENT DAVID NIEVES $185,842.41 POLICE OFFICER POLICE DEPARTMENT RALPH VALENZISI $185,823.47 CHIEF OF TECH/INN/PART INFORMATION TECHNOLOGY ANTHONY BRUNO $185,038.39 FIREFIGHTER FIRE DEPARTMENT GEORGE DALEY $184,219.06 POLICE OFFICER POLICE DEPARTMENT JOHN HAGGERTY $184,011.94 POLICE OFFICER POLICE DEPARTMENT SUZANNE KOROSHETZ $180,451.94 PRINCIPAL BRIEN MCMAHON HIGH SCHOOL REGINALD ROBERTS $179,428.49 PRINCIPAL NORWALK HIGH SCHOOL ANTHONY DITRIO $175,401.92 PRINCIPAL KENDALL ELEMENTARY SCHOOL JOHN REYNOLDS $174,651.99 PRINCIPAL JEFFERSON ELEMENTARY SCHOOL ASHLEY GONZALEZ $173,691.52 DEPUTY POLICE CHIEF POLICE DEPARTMENT MICHAEL SILVA $173,378.57 POLICE OFFICER POLICE DEPARTMENT THOMAS HAMILTON $172,760.39 DIRECTOR OF FINANCE FINANCE CITY LINDA SUMPTER $172,449.88 PRINCIPAL PONUS RIDGE MIDDLE SCHOOL MARK SUDA $171,022.82 POLICE OFFICER POLICE DEPARTMENT PAUL KRASNAVAGE $170,344.01 PRINCIPAL TRACEY ELEMENTARY SCHOOL PRAVEEN JOHN $169,551.67 POLICE LIEUTENANT POLICE DEPARTMENT LYNNE MOORE $169,366.08 PRINCIPAL WEST ROCKS MIDDLE SCHOOL CARL WILLIAMS $169,079.77 POLICE OFFICER POLICE DEPARTMENT ALBERT SACKEY $168,212.00 PRINCIPAL NATHAN HALE MIDDLE SCHOOL JOSEPH VELLUCCI $167,865.92 PRINCIPAL -
Commencement Program 2021
Sapientia et Doctrina Wisdom and Learning “Wisdom and learning shall be the stability of thy times.” —Adapted from Isaiah 33:6 The Fordham University Seal The Great Seal of Fordham University proclaims that Fordham has been a Jesuit university since its founder, Archbishop John Hughes, entrusted it to the care of the Society of Jesus five years after its founding in 1841. Hence, the coat of arms of the Society of Jesus stands at the center of the Great Seal of the University. The coat of arms bears the Greek letters for the name Jesus—IHS—with the cross resting in the horizontal line of the letter H, and the three nails beneath in a field framed in maroon, the color of the University, with fleurs-de-lis on the edge of the maroon frame. Around the Society’s coat of arms is a scroll with the University’s motto, Sapientia et Doctrina (Wisdom and Learning). The scroll rests on a field in which tongues of fire are displayed, recalling the outpouring of the gifts of the Holy Spirit of wisdom (sapientia) that marked the first Pentecost. A laurel wreath at the center of which are listed the names of the disciplines that are or have been taught at the University rests at the top of the seal. (The University had a medical school from 1905 to 1919 and a College of Pharmacy from 1912 to 1971.) These central heraldic devices are enclosed within a circular field fashioned as a belt and edged with beads. The field bears the University’s name (rendered in Latin) and the date of its foundation. -
Index to St. Louis, Missouri Naturalization Records Created After Sept
Index to St. Louis, Missouri Naturalization Records Created after Sept. 27, 1906 Alphabetical surname index N–R History & Genealogy Department St. Louis County Library 1640 S. Lindberg Blvd. St. Louis, Missouri 63131 314-994-3300, ext. 2070 [email protected] Index to St. Louis, Missouri Naturalization Records Created after Sept. 27, 1906 This index covers St. Louis, Missouri naturalization records created between October 1, 1906 and December 1928 and is based on the following sources: • Naturalizations, U.S. District Court—Eastern Division, Eastern Judicial District of Missouri, Vols. 1 – 82 • Naturalizations, U.S. Circuit Court— Eastern Division, Eastern Judicial District of Missouri, Vols. 5 – 21 Entries are listed alphabetically by surname, then by given name, and then numerically by volume number. Abbreviations and Notations SLCL = History and Genealogy Department microfilm number (St. Louis County Library) FHL = Family History Library microfilm number * = spelling taken from the signature which differed from name in index. How to obtain copies Photocopies of indexed articles may be requested by sending an email to the History and Genealogy Department at [email protected]. A limit of three searches per request applies. Please review the library's lookup policy at https://www.slcl.org/genealogy-and-local- history/services. A declaration of intention may lead to further records. For more information, contact the National Archives at the address below. Include all information listed on the declaration of intention. National Archives, Central Plains Region 400 W. Pershing Rd. Kansas City, MO 64108 (816) 268-8000 [email protected] History Genealogy Dept. Index to St. Louis, Missouri Naturalization Records St.