Developing a Non-Destructive Metabarcoding Protocol for Detection of Pest Insects in Bulk Trap Catches
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Plant Press
Special Symposium Issue continues on page 14 Department of Botany & the U.S. National Herbarium The Plant Press New Series - Vol. 20 - No. 3 July-September 2017 Botany Profile Plant Expeditions: History Has Its Eyes On You By Gary A. Krupnick he 15th Smithsonian Botani- as specimens (living or dried) in centuries field explorers to continue what they are cal Symposium was held at the past. doing. National Museum of Natural The symposium began with Laurence T he morning session began with a History (NMNH) and the U.S. Botanic Dorr (Chair of Botany, NMNH) giv- th Garden (USBG) on May 19, 2017. The ing opening remarks. Since the lectures series of talks focusing on the 18 symposium, titled “Exploring the Natural were taking place in Baird Auditorium, Tcentury explorations of Canada World: Plants, People and Places,” Dorr took the opportunity to talk about and the United States. Jacques Cayouette focused on the history of plant expedi- the theater’s namesake, Spencer Baird. A (Agriculture and Agri-Food Canada) tions. Over 200 participants gathered to naturalist, ornithologist, ichthyologist, and presented the first talk, “Moravian Mis- hear stories dedicated col- sionaries as Pioneers of Botanical Explo- and learn about lector, Baird was ration in Labrador (1765-1954).” He what moti- the first curator explained that missionaries of the Mora- vated botanical to be named vian Church, one of the oldest Protestant explorers of at the Smith- denominations, established missions the Western sonian Institu- along coastal Labrador in Canada in the Hemisphere in the 18th, 19th, and 20th tion and eventually served as Secretary late 1700s. -

Profile of Nancy A. Moran ‘‘ Always Liked Insects,’’ Says Nancy A
Profile of Nancy A. Moran ‘‘ always liked insects,’’ says Nancy A. bination,’’ she says. ‘‘Why have males Moran, Regent’s Professor of Ecol- and females, and not just reproduce by ogy and Evolutionary Biology at parthenogenesis and have all females?’’ the University of Arizona (Tuc- One of her advisors, William Hamilton, son,I AZ). ‘‘As a little kid, I was known had proposed that sexual reproduction as the girl who collected insects and had was important to create genetic diversity them in jars and things like that.’’ Years to stay one step ahead of coevolving later, this youthful bug collector has be- natural enemies, especially parasites and come a renowned entomologist whose pathogens. ‘‘I became interested in that work crosses over into multiple disci- idea and began looking at it in aphids,’’ plines, including microbiology, ecology, she says, ‘‘which are very useful since and molecular evolution. Moran’s re- they are parthogenetic for part of their search primarily focuses on the ecology life cycle’’ (3). and evolution of aphids and, since 1990, After receiving her Ph.D. in zoology has especially focused on the interaction in 1982, Moran spent the next several and coevolution of these small insects years studying evolutionary ecology in and the symbiotic bacteria that live in- aphids. ‘‘It was less than completely side of them. satisfying in a lot of ways,’’ she admits. ‘‘The whole evolution of insects has ‘‘At that time you were so far from the been in tandem with these bacteria,’’ actual genetic basis of the variation you Moran says. ‘‘We would not see insects were looking at, so you had no handle feeding on plant sap if it weren’t for as to which genes were actually causing symbiosis.’’ Elected to the National Nancy A. -

Diversity and Resource Choice of Flower-Visiting Insects in Relation to Pollen Nutritional Quality and Land Use
Diversity and resource choice of flower-visiting insects in relation to pollen nutritional quality and land use Diversität und Ressourcennutzung Blüten besuchender Insekten in Abhängigkeit von Pollenqualität und Landnutzung Vom Fachbereich Biologie der Technischen Universität Darmstadt zur Erlangung des akademischen Grades eines Doctor rerum naturalium genehmigte Dissertation von Dipl. Biologin Christiane Natalie Weiner aus Köln Berichterstatter (1. Referent): Prof. Dr. Nico Blüthgen Mitberichterstatter (2. Referent): Prof. Dr. Andreas Jürgens Tag der Einreichung: 26.02.2016 Tag der mündlichen Prüfung: 29.04.2016 Darmstadt 2016 D17 2 Ehrenwörtliche Erklärung Ich erkläre hiermit ehrenwörtlich, dass ich die vorliegende Arbeit entsprechend den Regeln guter wissenschaftlicher Praxis selbständig und ohne unzulässige Hilfe Dritter angefertigt habe. Sämtliche aus fremden Quellen direkt oder indirekt übernommene Gedanken sowie sämtliche von Anderen direkt oder indirekt übernommene Daten, Techniken und Materialien sind als solche kenntlich gemacht. Die Arbeit wurde bisher keiner anderen Hochschule zu Prüfungszwecken eingereicht. Osterholz-Scharmbeck, den 24.02.2016 3 4 My doctoral thesis is based on the following manuscripts: Weiner, C.N., Werner, M., Linsenmair, K.-E., Blüthgen, N. (2011): Land-use intensity in grasslands: changes in biodiversity, species composition and specialization in flower-visitor networks. Basic and Applied Ecology 12 (4), 292-299. Weiner, C.N., Werner, M., Linsenmair, K.-E., Blüthgen, N. (2014): Land-use impacts on plant-pollinator networks: interaction strength and specialization predict pollinator declines. Ecology 95, 466–474. Weiner, C.N., Werner, M , Blüthgen, N. (in prep.): Land-use intensification triggers diversity loss in pollination networks: Regional distinctions between three different German bioregions Weiner, C.N., Hilpert, A., Werner, M., Linsenmair, K.-E., Blüthgen, N. -

Old Woman Creek National Estuarine Research Reserve Management Plan 2011-2016
Old Woman Creek National Estuarine Research Reserve Management Plan 2011-2016 April 1981 Revised, May 1982 2nd revision, April 1983 3rd revision, December 1999 4th revision, May 2011 Prepared for U.S. Department of Commerce Ohio Department of Natural Resources National Oceanic and Atmospheric Administration Division of Wildlife Office of Ocean and Coastal Resource Management 2045 Morse Road, Bldg. G Estuarine Reserves Division Columbus, Ohio 1305 East West Highway 43229-6693 Silver Spring, MD 20910 This management plan has been developed in accordance with NOAA regulations, including all provisions for public involvement. It is consistent with the congressional intent of Section 315 of the Coastal Zone Management Act of 1972, as amended, and the provisions of the Ohio Coastal Management Program. OWC NERR Management Plan, 2011 - 2016 Acknowledgements This management plan was prepared by the staff and Advisory Council of the Old Woman Creek National Estuarine Research Reserve (OWC NERR), in collaboration with the Ohio Department of Natural Resources-Division of Wildlife. Participants in the planning process included: Manager, Frank Lopez; Research Coordinator, Dr. David Klarer; Coastal Training Program Coordinator, Heather Elmer; Education Coordinator, Ann Keefe; Education Specialist Phoebe Van Zoest; and Office Assistant, Gloria Pasterak. Other Reserve staff including Dick Boyer and Marje Bernhardt contributed their expertise to numerous planning meetings. The Reserve is grateful for the input and recommendations provided by members of the Old Woman Creek NERR Advisory Council. The Reserve is appreciative of the review, guidance, and council of Division of Wildlife Executive Administrator Dave Scott and the mapping expertise of Keith Lott and the late Steve Barry. -

The Genomics and Evolution of Mutualistic and Pathogenic Bacteria
Symbiotic bacteria in animals • Oct 3 2006 • Nancy Moran • Professor, Ecology and Evolutionary Biology Reading: The gut flora as a forgotten organ by A. O’Hara and F Shanahan EMBO Reports. 2006 What is symbiosis? • Term typically used for a chronic association of members of more than one genetic lineage, without overt pathogenesis • Often for mutual benefit, which may be easy or difficult to observe – Exchange of nutrients or other metabolic products, protection, transport, structural integrity Microbes in animal evolution • Bacteria present by 3.9 bya, Archaea and Eukaryota by >2 bya – The Earth is populated by ecologically diverse microbes • Animals appear about 1 bya • Animals evolved in microbial soup – “Innate” immune system probably universal among animal phyla: pathogenic infection was a constant selection pressure – But animals also evolved codependence on microbes, some of which are required for normal development and reproduction evolutionary innovations through symbiosis: examples • Eukaryotic cell (mitochondria) • Photosynthesis in eukaryotes (plastids) • Colonization of land by plants (mycorrhizae) • Nitrogen fixation by plants (rhizobia) • Animal life at deep sea vents (chemoautotrophic life systems) • Use of many nutrient-limited niches by animal lineages Why do hosts and symbionts cooperate so often? • Persistent association allows both to increase their persistence and replication. –Coinheritance – Long-term infection • Intimate metabolic exchange generating immediate beneficial feedback Symbiosis- main variables • Route -
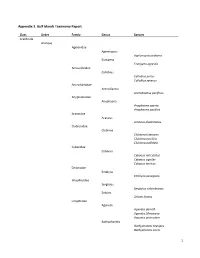
1 Appendix 3. Gulf Islands Taxonomy Report
Appendix 3. Gulf Islands Taxonomy Report Class Order Family Genus Species Arachnida Araneae Agelenidae Agelenopsis Agelenopsis utahana Eratigena Eratigena agrestis Amaurobiidae Callobius Callobius pictus Callobius severus Antrodiaetidae Antrodiaetus Antrodiaetus pacificus Anyphaenidae Anyphaena Anyphaena aperta Anyphaena pacifica Araneidae Araneus Araneus diadematus Clubionidae Clubiona Clubiona lutescens Clubiona pacifica Clubiona pallidula Cybaeidae Cybaeus Cybaeus reticulatus Cybaeus signifer Cybaeus tetricus Dictynidae Emblyna Emblyna peragrata Gnaphosidae Sergiolus Sergiolus columbianus Zelotes Zelotes fratris Linyphiidae Agyneta Agyneta darrelli Agyneta fillmorana Agyneta protrudens Bathyphantes Bathyphantes brevipes Bathyphantes keeni 1 Centromerita Centromerita bicolor Ceratinops Ceratinops latus Entelecara Entelecara acuminata Erigone Erigone aletris Erigone arctica Erigone cristatopalpus Frederickus Frederickus coylei Grammonota Grammonota kincaidi Linyphantes Linyphantes nehalem Linyphantes nigrescens Linyphantes pacificus Linyphantes pualla Linyphantes victoria Mermessus Mermessus trilobatus Microlinyphia Microlinyphia dana Neriene Neriene digna Neriene litigiosa Oedothorax Oedothorax alascensis Pityohyphantes Pityohyphantes alticeps Pocadicnemis Pocadicnemis pumila Poeciloneta Poeciloneta fructuosa Saaristoa Saaristoa sammamish Scotinotylus Scotinotylus sp. 5GAB Semljicola Semljicola sp. 1GAB Sisicottus Spirembolus Spirembolus abnormis Spirembolus mundus Tachygyna Tachygyna ursina Tachygyna vancouverana Tapinocyba Tapinocyba -

Insecta, Diptera) 213 Doi: 10.3897/Zookeys.441.7197 CHECKLIST Launched to Accelerate Biodiversity Research
A peer-reviewed open-access journal ZooKeys 441:Checklist 213–223 (2014) of the families Lonchopteridae and Phoridae of Finland (Insecta, Diptera) 213 doi: 10.3897/zookeys.441.7197 CHECKLIST www.zookeys.org Launched to accelerate biodiversity research Checklist of the families Lonchopteridae and Phoridae of Finland (Insecta, Diptera) Jere Kahanpää1 1 Finnish Museum of Natural History, Zoology Unit, P.O. Box 17, FI-00014 University of Helsinki, Finland Corresponding author: Jere Kahanpää ([email protected]) Academic editor: J. Salmela | Received 5 February 2014 | Accepted 26 March 2014 | Published 19 September 2014 http://zoobank.org/0C0D4F58-4F6C-488B-B3F0-ECFDF449FEF1 Citation: Kahanpää J (2014) Checklist of the families Lonchopteridae and Phoridae of Finland (Insecta, Diptera). In: Kahanpää J, Salmela J (Eds) Checklist of the Diptera of Finland. ZooKeys 441: 213–223. doi: 10.3897/zookeys.441.7197 Abstract A checklist of the Lonchopteridae and Phoridae recorded from Finland is presented. Keywords Checklist, Finland, Diptera, Lonchopteridae, Phoridae Introduction The superfamily Phoroidea includes at least the fly families Phoridae, Lonchopteridae, and the small family Ironomyiidae known only from Australia. The flat-footed fly families Platypezidae and Opetiidae, treated in a separate paper in this volume, are placed either in their own basal superfamily Platypezoidea or included in Phoroidea (see Cumming et al. 1995, Woodley et al. 2009 and Wiegmann et al. 2011). Lonchoptera Meigen, 1803 is the only currently recognized genus in Lonchopteri- dae. Five Lonchoptera species have been added to the Finnish fauna after the checklist of Hackman (1980) (Andersson 1991, Kahanpää 2013). The scuttle flies (family Phoridae) may be the largest single family in Diptera. -

The Importance of Vegetation Configuration in Coastal
Biology Department Research Group Terrestrial Ecology _____________________________________________________________________________________ THE IMPORTANCE OF VEGETATION CONFIGURATION IN COASTAL DUNES TO PRESERVE DIVERSITY OF MARRAM- ASSOCIATED INVERTEBRATES IS HABITAT CONFIGURATION A DRIVER OF DIVERSITY IN DUNES? Noëmie Van den Bon Studentnumber: 01506438 Supervisor(s): Prof. Dr. Dries Bonte Dr. Martijn Vandegehuchte Scientific tutor: Ruben Van De Walle Master’s dissertation submitted to obtain the degree of Master of Science in Biology Academic year: 2019 - 2020 © Faculty of Sciences – research group Terrestrial Ecology All rights reserved. This thesis contains confidential information and confidential research results that are property to the UGent. The contents of this master thesis may under no circumstances be made public, nor complete or partial, without the explicit and preceding permission of the UGent representative, i.e. the supervisor. The thesis may under no circumstances be copied or duplicated in any form, unless permission granted in written form. Any violation of the confidential nature of this thesis may impose irreparable damage to the UGent. In case of a dispute that may arise within the context of this declaration, the Judicial Court of Gent only is competent to be notified. 2 Table of content 1. Introduction ....................................................................................................................................... 5 1.1. The status of biodiversity and ecosystems .......................................................................................... -

Observations on Antennal Morphology in Diptera, with Particular Reference to the Articular Surfaces Between Segments 2 and 3 in the Cyclorrhapha
© The Author, 2011. Journal compilation © Australian Museum, Sydney, 2011 Records of the Australian Museum (2011) Vol. 63: 113–166. ISSN 0067-1975 doi:10.3853/j.0067-1975.63.2011.1585 Observations on Antennal Morphology in Diptera, with Particular Reference to the Articular Surfaces between Segments 2 and 3 in the Cyclorrhapha DaviD K. Mcalpine Australian Museum, 6 College Street, Sydney NSW 2010, Australia abstract. The main features of antennal segments 2 and 3 seen in the higher Diptera are described, including many that are not or inadequately covered in available publications. The following terms are introduced or clarified: for segment 2 or the pedicel—annular ridge, caestus, chin, collar, conus, distal articular surface, encircling furrow, foramen of articulation, foraminal cusp, foraminal ring, pedicellar button, pedicellar cup, rim; for segment 3 or the postpedicel—basal foramen, basal hollow, basal stem, postpedicellar pouch, sacculus, scabrous tongue, sub-basal caecum; for the stylus or arista—stylar goblet. Particular attention is given to the occurrence and position of the pedicellar button. The button is the cuticular component of a chordotonal organ, which perhaps has the role of a baroreceptor. It is present in the majority of families of Diptera, and possibly was present in the ancestral dipteran. Some generalizations about antennal structure are made, and a diagram showing the main trends in antennal evolution in the Eremoneura is provided. The general form of the antenna shows a transition from approximate radial symmetry (e.g., in Empis, Microphor, and Opetia) through to superficial bilateral symmetry (in many taxa of Eumuscomorpha), though there is usually much asymmetry in detail. -

ABSTRACT BAYLESS, KEITH MOHR. Phylogenomic Studies of Evolutionary Radiations of Diptera
ABSTRACT BAYLESS, KEITH MOHR. Phylogenomic Studies of Evolutionary Radiations of Diptera. (Under the direction of Dr. Brian M. Wiegmann.) Efforts to understand the evolutionary history of flies have been obstructed by the lack of resolution in major radiations. Diptera is a highly diverse branch on the tree of life, but this diversity accrued at an uneven pace. Some of radiations that contributed disproportionately to species diversity occurred contemporaneously, and understanding the relationships of these taxa can illuminate broad scale patterns. Relationships between some subordinate groups of taxa are notoriously difficult to untangle, and genomic data will address these problems at a new scale. This project will focus on two major radiations in Diptera: Tabanus horse flies and relatives, and acalyptrate Schizophora. Tabanus includes over one thousand species. Synthesis focused research on the group is stymied by its species richness, worldwide distribution, inconsistent diagnosis, and scale of undescribed diversity. Furthermore, the genus may be non-monophyletic with respect to more than 10 other lineages of horse flies. A groundwork phylogenetic study of worldwide Tabanus is needed to understand the evolution of this lineage and to make comprehensive taxonomic projects manageable. Data to address this question was collected from two different sources. A dataset including five genes was sequenced from ninety-four species in the Tabanus group, including nearly all genera of Tabanini and at least one species from every biogeographic region. Then a new data source from a next generation sequencing approach, Anchored Hybrid Enrichment exome capture, was used to accumulate a dataset including hundreds of genes for a subset of the taxa. -

Final Report 2006
Fly Assemblages of Sandy Exposed Riverine Sediment Final Report - April 2007 John Spedan Lewis Foundation C. M. Drake, A. Godfrey, S. M. Hewitt & J. Parker Martin DRAKE Orchid House, Burridge, Axminster, Devon EX13 7DF [email protected] ANDY GODFREY 90 Bence Lane, Darton, Barnsley, South Yorkshire S75 5DA [email protected] STEPHEN M. HEWITT Tullie House, Castle Street, Carlisle, Cumbria CA3 8TP [email protected] JOHN PARKER 16 Brunswick Road, Penrith, Cumbria CA11 7LT 2 Summary This report describes surveys of flies at sandy exposed riverine sediment and forms part of a series of projects based on the flies included in the Biodiversity Action Plan. Eighteen rivers in England, Wales and Scotland were selected for survey on the basis of their supporting populations of the BPS-listed Spiriverpa lunulata , Cliorismia rustica or Rhabdomastix ‘laeta’ , or whose sandy aspect suggested that high potential for these species. Therevids were sought during unstructured searches lasting for about an hour. The assemblage of flies using ERS was sampled using timed sweep-netting at 284 points and suction sampling at 136 of these. Sampling was targeted at ERS rather than other riverine habitats. Species in all well represented families except sphaerocerids were identified. Records of therevids were patchy. New sites were found for Cliorismia on the Wey, Rother and Tay, and a previously known Rother population was found to be thriving. New records for Spiriverpa were from the Lune, Coquet, Till and Breamish. Cliorismia was found new to Scotland at two locations on the Tay and Spiriverpa was found to be widespread and locally numerous on both the Spey and the Tay (including several apparently new populations). -

Discovery of Lebambromyia in Myanmar Cretaceous Amber: Phylogenetic and Biogeographic Implications (Insecta, Diptera, Phoroidea)
insects Article Discovery of Lebambromyia in Myanmar Cretaceous Amber: Phylogenetic and Biogeographic Implications (Insecta, Diptera, Phoroidea) Davide Badano 1,2,* , Qingqing Zhang 3,4 , Michela Fratini 5, Laura Maugeri 5, Inna Bukreeva 5,6, Elena Longo 7 , Fabian Wilde 7 , David K. Yeates 8 and Pierfilippo Cerretti 1,2,8,* 1 Department of Biology and Biotechnologies “Charles Darwin”, Sapienza University of Rome, Piazzale A. Moro 5, 00185 Rome, Italy 2 Museum of Zoology, Sapienza University of Rome, Piazzale Valerio Massimo 6, 00162 Rome, Italy 3 State Key Laboratory of Palaeobiology and Stratigraphy, Nanjing Institute of Geology and Palaeontology and Center for Excellence in Life and Paleoenvironment, Chinese Academy of Sciences, 39 East Beijing Road, Nanjing 210008, China; [email protected] 4 Institute of Geosciences, University of Bonn, 53115 Bonn, Germany 5 CNR-Nanotec (Rome Unit) c/o Department of Physics, Sapienza University of Rome, Piazzale A. Moro, 5, 00185 Rome, Italy; [email protected] (M.F.); [email protected] (L.M.); [email protected] (I.B.) 6 P. N. Lebedev Physical Institute, Russian Academy of Sciences, Leninskii pr., 119991 Moscow, Russia 7 Helmholtz-Zentrum Geesthacht, Institute of Materials Physics, Max-Planck-Strasse 1, 21502 Geesthacht, Germany; [email protected] (E.L.); [email protected] (F.W.) 8 Australian National Insect Collection, CSIRO National Facilities and Collections, Black Mountain, Clunies Ross Street, Acton, Canberra, ACT 2601, Australia; [email protected] Citation: Badano, D.; Zhang, Q.; * Correspondence: [email protected] (D.B.); pierfi[email protected] (P.C.) Fratini, M.; Maugeri, L.; Bukreeva, I.; Longo, E.; Wilde, F.; Yeates, D.K.; Simple Summary: Phoroid flies are an ancient lineage of Diptera, which includes megadiverse, Cerretti, P.