202067Orig1s000
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Preventive Report Appendix
Title Authors Published Journal Volume Issue Pages DOI Final Status Exclusion Reason Nasal sumatriptan is effective in treatment of migraine attacks in children: A Ahonen K.; Hamalainen ML.; Rantala H.; 2004 Neurology 62 6 883-7 10.1212/01.wnl.0000115105.05966.a7 Deemed irrelevant in initial screening Seasonal variation in migraine. Alstadhaug KB.; Salvesen R.; Bekkelund SI. Cephalalgia : an 2005 international journal 25 10 811-6 10.1111/j.1468-2982.2005.01018.x Deemed irrelevant in initial screening Flunarizine, a calcium channel blocker: a new prophylactic drug in migraine. Amery WK. 1983 Headache 23 2 70-4 10.1111/j.1526-4610.1983.hed2302070 Deemed irrelevant in initial screening Monoamine oxidase inhibitors in the control of migraine. Anthony M.; Lance JW. Proceedings of the 1970 Australian 7 45-7 Deemed irrelevant in initial screening Prostaglandins and prostaglandin receptor antagonism in migraine. Antonova M. 2013 Danish medical 60 5 B4635 Deemed irrelevant in initial screening Divalproex extended-release in adolescent migraine prophylaxis: results of a Apostol G.; Cady RK.; Laforet GA.; Robieson randomized, double-blind, placebo-controlled study. WZ.; Olson E.; Abi-Saab WM.; Saltarelli M. 2008 Headache 48 7 1012-25 10.1111/j.1526-4610.2008.01081.x Deemed irrelevant in initial screening Divalproex sodium extended-release for the prophylaxis of migraine headache in Apostol G.; Lewis DW.; Laforet GA.; adolescents: results of a stand-alone, long-term open-label safety study. Robieson WZ.; Fugate JM.; Abi-Saab WM.; 2009 Headache 49 1 45-53 10.1111/j.1526-4610.2008.01279.x Deemed irrelevant in initial screening Safety and tolerability of divalproex sodium extended-release in the prophylaxis of Apostol G.; Pakalnis A.; Laforet GA.; migraine headaches: results of an open-label extension trial in adolescents. -

8Th European Congress on Epileptology, Berlin, Germany, 21 – 25 September 2008
Epilepsia, 50(Suppl. 4): 2–262, 2009 doi: 10.1111/j.1528-1167.2009.02063.x 8th ECE PROCEEDINGS 8th European Congress on Epileptology, Berlin, Germany, 21 – 25 September 2008 Sunday 21 September 2008 KV7 channels (KV7.1-5) are encoded by five genes (KCNQ1-5). They have been identified in the last 10–15 years by discovering the caus- 14:30 – 16:00 ative genes for three autosomal dominant diseases: cardiac arrhythmia Hall 1 (long QT syndrome, KCNQ1), congenital deafness (KCNQ1 and KCNQ4), benign familial neonatal seizures (BFNS, KCNQ2 and VALEANT PHARMACEUTICALS SATELLITE SYM- KCNQ3), and peripheral nerve hyperexcitability (PNH, KCNQ2). The fifth member of this gene family (KCNQ5) is not affected in a disease so POSIUM – NEURON-SPECIFIC M-CURRENT K+ CHAN- far. The phenotypic spectrum associated with KCNQ2 mutations is prob- NELS: A NEW TARGET IN MANAGING EPILEPSY ably broader than initially thought (i.e. not only BFNS), as patients with E. Perucca severe epilepsies and developmental delay, or with Rolando epilepsy University of Pavia, Italy have been described. With regard to the underlying molecular pathophys- iology, it has been shown that mutations in KCNQ2 and KCNQ3 Innovations in protein biology, coupled with genetic manipulations, have decrease the resulting K+ current thereby explaining the occurrence of defined the structure and function of many of the voltage- and ligand- epileptic seizures by membrane depolarization and increased neuronal gated ion channels, channel subunits, and receptors that are the underpin- firing. Very subtle changes restricted to subthreshold voltages are suffi- nings of neuronal hyperexcitability and epilepsy. Of the currently cient to cause BFNS which proves in a human disease model that this is available antiepileptic drugs (AEDs), no two act in the same way, but all the relevant voltage range for these channels to modulate the firing rate. -

PR2 2009.Vp:Corelventura
Pharmacological Reports Copyright © 2009 2009, 61, 197216 by Institute of Pharmacology ISSN 1734-1140 Polish Academy of Sciences Review Third-generation antiepileptic drugs: mechanisms of action, pharmacokinetics and interactions Jarogniew J. £uszczki1,2 Department of Pathophysiology, Medical University of Lublin, Jaczewskiego 8, PL 20-090 Lublin, Poland Department of Physiopathology, Institute of Agricultural Medicine, Jaczewskiego 2, PL 20-950 Lublin, Poland Correspondence: Jarogniew J. £uszczki, e-mail: [email protected]; [email protected] Abstract: This review briefly summarizes the information on the molecular mechanisms of action, pharmacokinetic profiles and drug interac- tions of novel (third-generation) antiepileptic drugs, including brivaracetam, carabersat, carisbamate, DP-valproic acid, eslicar- bazepine, fluorofelbamate, fosphenytoin, ganaxolone, lacosamide, losigamone, pregabalin, remacemide, retigabine, rufinamide, safinamide, seletracetam, soretolide, stiripentol, talampanel, and valrocemide. These novel antiepileptic drugs undergo intensive clinical investigations to assess their efficacy and usefulness in the treatment of patients with refractory epilepsy. Key words: antiepileptic drugs, brivaracetam, carabersat, carisbamate, DP-valproic acid, drug interactions, eslicarbazepine, fluorofelbamate, fosphenytoin, ganaxolone, lacosamide, losigamone, pharmacokinetics, pregabalin, remacemide, retigabine, rufinamide, safinamide, seletracetam, soretolide, stiripentol, talampanel, valrocemide Abbreviations: 4-AP -

A Comparative Effectiveness Meta-Analysis of Drugs for the Prophylaxis of Migraine Headache
University of South Florida Masthead Logo Scholar Commons School of Information Faculty Publications School of Information 7-2015 A Comparative Effectiveness Meta-Analysis of Drugs for the Prophylaxis of Migraine Headache Authors: Jeffrey L. Jackson, Elizabeth Cogbill, Rafael Santana-Davila, Christina Eldredge, William Collier, Andrew Gradall, Neha Sehgal, and Jessica Kuester OBJECTIVE: To compare the effectiveness and side effects of migraine prophylactic medications. DESIGN: We performed a network meta-analysis. Data were extracted independently in duplicate and quality was assessed using both the JADAD and Cochrane Risk of Bias instruments. Data were pooled and network meta-analysis performed using random effects models. DATA SOURCES: PUBMED, EMBASE, Cochrane Trial Registry, bibliography of retrieved articles through 18 May 2014. ELIGIBILITY CRITERIA FOR SELECTING STUDIES: We included randomized controlled trials of adults with migraine headaches of at least 4 weeks in duration. RESULTS: Placebo controlled trials included alpha blockers (n = 9), angiotensin converting enzyme inhibitors (n = 3), angiotensin receptor blockers (n = 3), anticonvulsants (n = 32), beta-blockers (n = 39), calcium channel blockers (n = 12), flunarizine (n = 7), serotonin reuptake inhibitors (n = 6), serotonin norepinephrine reuptake inhibitors (n = 1) serotonin agonists (n = 9) and tricyclic antidepressants (n = 11). In addition there were 53 trials comparing different drugs. Drugs with at least 3 trials that were more effective than placebo for episodic migraines -

Molecular Mechanisms of Antiseizure Drug Activity at GABAA Receptors
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Seizure 22 (2013) 589–600 Contents lists available at SciVerse ScienceDirect Seizure jou rnal homepage: www.elsevier.com/locate/yseiz Review Molecular mechanisms of antiseizure drug activity at GABAA receptors L. John Greenfield Jr.* Dept. of Neurology, University of Arkansas for Medical Sciences, 4301W. Markham St., Slot 500, Little Rock, AR 72205, United States A R T I C L E I N F O A B S T R A C T Article history: The GABAA receptor (GABAAR) is a major target of antiseizure drugs (ASDs). A variety of agents that act at Received 6 February 2013 GABAARs s are used to terminate or prevent seizures. Many act at distinct receptor sites determined by Received in revised form 16 April 2013 the subunit composition of the holoreceptor. For the benzodiazepines, barbiturates, and loreclezole, Accepted 17 April 2013 actions at the GABAAR are the primary or only known mechanism of antiseizure action. For topiramate, felbamate, retigabine, losigamone and stiripentol, GABAAR modulation is one of several possible Keywords: antiseizure mechanisms. Allopregnanolone, a progesterone metabolite that enhances GABAAR function, Inhibition led to the development of ganaxolone. Other agents modulate GABAergic ‘‘tone’’ by regulating the Epilepsy synthesis, transport or breakdown of GABA. GABAAR efficacy is also affected by the transmembrane Antiepileptic drugs chloride gradient, which changes during development and in chronic epilepsy. This may provide an GABA receptor Seizures additional target for ‘‘GABAergic’’ ASDs. GABAAR subunit changes occur both acutely during status Chloride channel epilepticus and in chronic epilepsy, which alter both intrinsic GABAAR function and the response to GABAAR-acting ASDs. -

Rat Animal Models for Screening Medications to Treat Alcohol Use Disorders
ACCEPTED MANUSCRIPT Selectively Bred Rats Page 1 of 75 Rat Animal Models for Screening Medications to Treat Alcohol Use Disorders Richard L. Bell*1, Sheketha R. Hauser1, Tiebing Liang2, Youssef Sari3, Antoinette Maldonado-Devincci4, and Zachary A. Rodd1 1Indiana University School of Medicine, Department of Psychiatry, Indianapolis, IN 46202, USA 2Indiana University School of Medicine, Department of Gastroenterology, Indianapolis, IN 46202, USA 3University of Toledo, Department of Pharmacology, Toledo, OH 43614, USA 4North Carolina A&T University, Department of Psychology, Greensboro, NC 27411, USA *Send correspondence to: Richard L. Bell, Ph.D.; Associate Professor; Department of Psychiatry; Indiana University School of Medicine; Neuroscience Research Building, NB300C; 320 West 15th Street; Indianapolis, IN 46202; e-mail: [email protected] MANUSCRIPT Key Words: alcohol use disorder; alcoholism; genetically predisposed; selectively bred; pharmacotherapy; family history positive; AA; HAD; P; msP; sP; UChB; WHP Chemical compounds studied in this article Ethanol (PubChem CID: 702); Acamprosate (PubChem CID: 71158); Baclofen (PubChem CID: 2284); Ceftriaxone (PubChem CID: 5479530); Fluoxetine (PubChem CID: 3386); Naltrexone (PubChem CID: 5360515); Prazosin (PubChem CID: 4893); Rolipram (PubChem CID: 5092); Topiramate (PubChem CID: 5284627); Varenicline (PubChem CID: 5310966) ACCEPTED _________________________________________________________________________________ This is the author's manuscript of the article published in final edited form as: Bell, R. L., Hauser, S. R., Liang, T., Sari, Y., Maldonado-Devincci, A., & Rodd, Z. A. (2017). Rat animal models for screening medications to treat alcohol use disorders. Neuropharmacology. https://doi.org/10.1016/j.neuropharm.2017.02.004 ACCEPTED MANUSCRIPT Selectively Bred Rats Page 2 of 75 The purpose of this review is to present animal research models that can be used to screen and/or repurpose medications for the treatment of alcohol abuse and dependence. -

Patent Application Publication ( 10 ) Pub . No . : US 2019 / 0192440 A1
US 20190192440A1 (19 ) United States (12 ) Patent Application Publication ( 10) Pub . No. : US 2019 /0192440 A1 LI (43 ) Pub . Date : Jun . 27 , 2019 ( 54 ) ORAL DRUG DOSAGE FORM COMPRISING Publication Classification DRUG IN THE FORM OF NANOPARTICLES (51 ) Int . CI. A61K 9 / 20 (2006 .01 ) ( 71 ) Applicant: Triastek , Inc. , Nanjing ( CN ) A61K 9 /00 ( 2006 . 01) A61K 31/ 192 ( 2006 .01 ) (72 ) Inventor : Xiaoling LI , Dublin , CA (US ) A61K 9 / 24 ( 2006 .01 ) ( 52 ) U . S . CI. ( 21 ) Appl. No. : 16 /289 ,499 CPC . .. .. A61K 9 /2031 (2013 . 01 ) ; A61K 9 /0065 ( 22 ) Filed : Feb . 28 , 2019 (2013 .01 ) ; A61K 9 / 209 ( 2013 .01 ) ; A61K 9 /2027 ( 2013 .01 ) ; A61K 31/ 192 ( 2013. 01 ) ; Related U . S . Application Data A61K 9 /2072 ( 2013 .01 ) (63 ) Continuation of application No. 16 /028 ,305 , filed on Jul. 5 , 2018 , now Pat . No . 10 , 258 ,575 , which is a (57 ) ABSTRACT continuation of application No . 15 / 173 ,596 , filed on The present disclosure provides a stable solid pharmaceuti Jun . 3 , 2016 . cal dosage form for oral administration . The dosage form (60 ) Provisional application No . 62 /313 ,092 , filed on Mar. includes a substrate that forms at least one compartment and 24 , 2016 , provisional application No . 62 / 296 , 087 , a drug content loaded into the compartment. The dosage filed on Feb . 17 , 2016 , provisional application No . form is so designed that the active pharmaceutical ingredient 62 / 170, 645 , filed on Jun . 3 , 2015 . of the drug content is released in a controlled manner. Patent Application Publication Jun . 27 , 2019 Sheet 1 of 20 US 2019 /0192440 A1 FIG . -

Drug Class Review Newer Anticonvulsant Agents 28:12:92 Anticonvulsants, Other
Drug Class Review Newer Anticonvulsant Agents 28:12:92 Anticonvulsants, Other Brivaracetam (Briviact®) Clobazam (Onfi®) Eslicarbazepine (Aptiom®) Ezogabine (Potiga®) Felbamate (Felbatol®, others) Gabapentin (Neurontin®) Lacosamide (Vimpat®, others) Lamotrigine (Lamictal®, others) Levetiracetam (Keppra®, others) Oxcarbazepine (Trileptal®, Oxtellar XR®, others) Perampanel (Fycompa®) Pregabalin (Lyrica ®) Rufinamide (Banzel®) Tiagabine (Gabitril®) Topiramate (Topamax ®, Trokendi XR, Qudenxi XR, others) Vigabatrin (Sabril®) Zonisamide (Zonergan ®, others) Final Report June 2016 Review prepared by: Vicki Frydrych, Clinical Pharmacist University of Utah College of Pharmacy Copyright © 2016 by University of Utah College of Pharmacy Salt Lake City, Utah. All rights reserved. Table of Contents Introduction ....................................................................................................................... 1 Table 1: Comparison of Newer Anticonvulsant Agents ...................................... 2 Table 2: FDA-Approved Indications for Newer Anticonvulsant Agents ........... 16 Disease Overview ............................................................................................................ 17 Table 3: The International League Against Epilepsy Classification of Seizures ............................................................................................... 19 Table 4: Newer Antiepileptic Drugs Which May Exacerbate Seizures ............. 21 Table 5: Clinical Practice Guideline Recommendations for Epilepsy ............. -

(Eicah): Anti-Seizure Drugs Robert S. Fisher, MD
Epilepsy iConnect at Home (EiCaH): Anti-Seizure Drugs by Robert S. Fisher, MD, PhD Professor and Director, Stanford Epilepsy Center Copyright 2016 (use permission to Epilepsy Foundation of Northern California) This is a discussion about drugs to treat epilepsy. The big categories of how we treat epilepsy are with medicines, surgery, ketogenic diet, neurostimulation (such as the vagus nerve stimulation), biofeedback, and alternative therapies. They are all potential options, but by far the mainstay of treatment and the most used therapy is with anti-seizure medications. That will be the focus on what I talk about today. How well do anti-seizure medicines work? In general, about two-thirds of people who take anti-seizure medicines have their seizures controlled – meaning they have no seizures or very rare breakthrough seizures under unusual circumstances. And then one-third take medicine, but their seizures are uncontrolled either because of continued seizures or inability to take medications due to side-effects. That’s still a pretty large group of people, considering that 1% of the world has epilepsy. One-third of 1% is a number larger than all brain tumors, larger than all multiple sclerosis, larger than all muscular dystrophy. Epilepsy is not yet a solved problem. We have many anti-seizure drugs. Typically, you’ll hear them called AEDs – anti-epileptic drugs. But that’s a misnomer. None of these drugs cure epilepsy, so they’re not really anti-epilepsy drugs, they are seizure suppressant drugs. When you stop the medication, the epilepsy, which is the tendency to have recurrent seizures, remains. -

When to Start and How to Select Aeds
When to start & How to select AEDs Pasiri Sithinamsuwan, MD Division of Neurology, Phramongkutklao Hospital Concepts of medical treatments Treatment Initiation Consider other options if seizures are provoked Balance risks between recurrent seizures and adverse events of AEDs Frequency of seizures and risk of recurrent Psychosocial consequences of further seizures Avoid AEDs when diagnosis is in doubt AED do not prevent development of epilepsy Expectations should be modest (50%) First seizure, evaluate high recurrence risk A very high risk of recurrence Examples A single seizure occurring at least a month after a stroke A child with a single seizure conjoined with a structural or remote symptomatic etiology and an epileptiform EEG study A patient in whom diagnosis of a specific epilepsy syndrome associated with persistent threshold alteration can be made after the occurrence of a single seizure A first seizure might present present as status epilepticus ILAE 2014 Consider (a case-by-case basis) Seizure type, syndromic form Patient characteristics; age, gender. comorbidities Efficacy and side effect profile Dosing schedule, drug interaction Medical expertise Cost, ED drug (national formulary) Ideal properties for an easy-to-use antiepileptic drug Broad spectrum No tolerance High efficacy No withdrawal seizures Good tolerability Favorable pharmacokinetics (linear kinetics, T1/2 for 1-2 daily dosing) No risk of allergic or idiosyncratic reactions including teratogenicity Fast and easy dose escalation rate Low interaction -
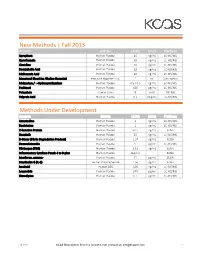
New Methods | Fall 2013 Methods Under Development
New Methods | Fall 2013 Analyte Matrix LLOQ Units Platform Aprepitant Human Plasma 10 ng/mL LC-MS/MS Ciprofloxacin Human Plasma 10 ng/mL LC-MS/MS Clonidine Human Plasma 10 pg/mL LC-MS/MS Deoxycholic Acid Human Plasma 10 ng/mL LC-MS/MS Mefenamic Acid Human Plasma 40 ng/mL LC-MS/MS Menstrual Blood (as Alkaline Hematin) Feminine Hygiene Prod. 1 mL Colorimetric Midazolam / α-Hydroxymidazolam Human Plasma 0.5 / 0.5 ng/mL LC-MS/MS Paclitaxel Human Plasma 100 pg/mL LC-MS/MS Potassium Human Urine 8 mM ICP-MS Valproic Acid Human Plasma 0.5 mcg/mL LC-MS/MS Methods Under Development Analyte Matrix LLOQ Units Platform Amantadine Human Plasma 2 ng/mL LC-MS/MS Bupivicaine Human Plasma 1 ng/mL LC-MS/MS C-Reactive Protein Human Plasma 62.5 ng/mL ELISA Dasatinib Human Plasma 20 ng/mL LC-MS/MS D-Dimer (Fibrin Degradation Product) Human Plasma 1.37 ng/mL ELISA Dexmedetomide Human Plasma 5 pg/mL LC-MS/MS Fibrinogen (FBG) Human Plasma 3.13 ng/mL ELISA Inflammatory Cytokine Panel: 4 to 9-plex Human Plasma Inquire ELISA Interferon, gamma- Human Plasma 15 pg/mL ELISA Interleukin-6 (IL-6) Human Plasma/Serum 1.56 pg/mL ELISA Isoniazid Human DBS 100 ng/mL LC-MS/MS Leuprolide Human Plasma 100 pg/ml LC-MS/MS Nimodipine Human Plasma 0.5 ng/mL LC-MS/MS Fall 2013 KCAS Bioanalytical Services | kcasbio.com | contact us: [email protected] 1 Analgesic Methods Analyte Matrix LLOQ Units Platform Acetaminophen (APAP) Human Plasma 0.1 mcg/mL LC-MS/MS Acetaminophen / Tramadol Human Plasma 100 / 5 ng/mL LC-MS/MS Acetylsalicylic Acid / Salicylic Acid Human Plasma 0.25 / 0.75 mcg/mL -

Stembook 2018.Pdf
The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances FORMER DOCUMENT NUMBER: WHO/PHARM S/NOM 15 WHO/EMP/RHT/TSN/2018.1 © World Health Organization 2018 Some rights reserved. This work is available under the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 IGO licence (CC BY-NC-SA 3.0 IGO; https://creativecommons.org/licenses/by-nc-sa/3.0/igo). Under the terms of this licence, you may copy, redistribute and adapt the work for non-commercial purposes, provided the work is appropriately cited, as indicated below. In any use of this work, there should be no suggestion that WHO endorses any specific organization, products or services. The use of the WHO logo is not permitted. If you adapt the work, then you must license your work under the same or equivalent Creative Commons licence. If you create a translation of this work, you should add the following disclaimer along with the suggested citation: “This translation was not created by the World Health Organization (WHO). WHO is not responsible for the content or accuracy of this translation. The original English edition shall be the binding and authentic edition”. Any mediation relating to disputes arising under the licence shall be conducted in accordance with the mediation rules of the World Intellectual Property Organization. Suggested citation. The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances. Geneva: World Health Organization; 2018 (WHO/EMP/RHT/TSN/2018.1). Licence: CC BY-NC-SA 3.0 IGO. Cataloguing-in-Publication (CIP) data.