Ecdysozoan Phylogeny and Bayesian Inference: First Use of Nearly Complete 28S and 18S Rrna Gene Sequences to Classify the Arthro
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The 2014 Golden Gate National Parks Bioblitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event
National Park Service U.S. Department of the Interior Natural Resource Stewardship and Science The 2014 Golden Gate National Parks BioBlitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event Natural Resource Report NPS/GOGA/NRR—2016/1147 ON THIS PAGE Photograph of BioBlitz participants conducting data entry into iNaturalist. Photograph courtesy of the National Park Service. ON THE COVER Photograph of BioBlitz participants collecting aquatic species data in the Presidio of San Francisco. Photograph courtesy of National Park Service. The 2014 Golden Gate National Parks BioBlitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event Natural Resource Report NPS/GOGA/NRR—2016/1147 Elizabeth Edson1, Michelle O’Herron1, Alison Forrestel2, Daniel George3 1Golden Gate Parks Conservancy Building 201 Fort Mason San Francisco, CA 94129 2National Park Service. Golden Gate National Recreation Area Fort Cronkhite, Bldg. 1061 Sausalito, CA 94965 3National Park Service. San Francisco Bay Area Network Inventory & Monitoring Program Manager Fort Cronkhite, Bldg. 1063 Sausalito, CA 94965 March 2016 U.S. Department of the Interior National Park Service Natural Resource Stewardship and Science Fort Collins, Colorado The National Park Service, Natural Resource Stewardship and Science office in Fort Collins, Colorado, publishes a range of reports that address natural resource topics. These reports are of interest and applicability to a broad audience in the National Park Service and others in natural resource management, including scientists, conservation and environmental constituencies, and the public. The Natural Resource Report Series is used to disseminate comprehensive information and analysis about natural resources and related topics concerning lands managed by the National Park Service. -

Investgating Determinants of Phylogeneic Accuracy
IMPACT OF MOLECULAR EVOLUTIONARY FOOTPRINTS ON PHYLOGENETIC ACCURACY – A SIMULATION STUDY Dissertation Submitted to The College of Arts and Sciences of the UNIVERSITY OF DAYTON In Partial Fulfillment of the Requirements for The Degree Doctor of Philosophy in Biology by Bhakti Dwivedi UNIVERSITY OF DAYTON August, 2009 i APPROVED BY: _________________________ Gadagkar, R. Sudhindra Ph.D. Major Advisor _________________________ Robinson, Jayne Ph.D. Committee Member Chair Department of Biology _________________________ Nielsen, R. Mark Ph.D. Committee Member _________________________ Rowe, J. John Ph.D. Committee Member _________________________ Goldman, Dan Ph.D. Committee Member ii ABSTRACT IMPACT OF MOLECULAR EVOLUTIONARY FOOTPRINTS ON PHYLOGENETIC ACCURACY – A SIMULATION STUDY Dwivedi Bhakti University of Dayton Advisor: Dr. Sudhindra R. Gadagkar An accurately inferred phylogeny is important to the study of molecular evolution. Factors impacting the accuracy of a phylogenetic tree can be traced to several consecutive steps leading to the inference of the phylogeny. In this simulation-based study our focus is on the impact of the certain evolutionary features of the nucleotide sequences themselves in the alignment rather than any source of error during the process of sequence alignment or due to the choice of the method of phylogenetic inference. Nucleotide sequences can be characterized by summary statistics such as sequence length and base composition. When two or more such sequences need to be compared to each other (as in an alignment prior to phylogenetic analysis) additional evolutionary features come into play, such as the overall rate of nucleotide substitution, the ratio of two specific instantaneous, rates of substitution (rate at which transitions and transversions occur), and the shape parameter, of the gamma distribution (that quantifies the extent of iii heterogeneity in substitution rate among sites in an alignment). -

Platyhelminthes, Nemertea, and "Aschelminthes" - A
BIOLOGICAL SCIENCE FUNDAMENTALS AND SYSTEMATICS – Vol. III - Platyhelminthes, Nemertea, and "Aschelminthes" - A. Schmidt-Rhaesa PLATYHELMINTHES, NEMERTEA, AND “ASCHELMINTHES” A. Schmidt-Rhaesa University of Bielefeld, Germany Keywords: Platyhelminthes, Nemertea, Gnathifera, Gnathostomulida, Micrognathozoa, Rotifera, Acanthocephala, Cycliophora, Nemathelminthes, Gastrotricha, Nematoda, Nematomorpha, Priapulida, Kinorhyncha, Loricifera Contents 1. Introduction 2. General Morphology 3. Platyhelminthes, the Flatworms 4. Nemertea (Nemertini), the Ribbon Worms 5. “Aschelminthes” 5.1. Gnathifera 5.1.1. Gnathostomulida 5.1.2. Micrognathozoa (Limnognathia maerski) 5.1.3. Rotifera 5.1.4. Acanthocephala 5.1.5. Cycliophora (Symbion pandora) 5.2. Nemathelminthes 5.2.1. Gastrotricha 5.2.2. Nematoda, the Roundworms 5.2.3. Nematomorpha, the Horsehair Worms 5.2.4. Priapulida 5.2.5. Kinorhyncha 5.2.6. Loricifera Acknowledgements Glossary Bibliography Biographical Sketch Summary UNESCO – EOLSS This chapter provides information on several basal bilaterian groups: flatworms, nemerteans, Gnathifera,SAMPLE and Nemathelminthes. CHAPTERS These include species-rich taxa such as Nematoda and Platyhelminthes, and as taxa with few or even only one species, such as Micrognathozoa (Limnognathia maerski) and Cycliophora (Symbion pandora). All Acanthocephala and subgroups of Platyhelminthes and Nematoda, are parasites that often exhibit complex life cycles. Most of the taxa described are marine, but some have also invaded freshwater or the terrestrial environment. “Aschelminthes” are not a natural group, instead, two taxa have been recognized that were earlier summarized under this name. Gnathifera include taxa with a conspicuous jaw apparatus such as Gnathostomulida, Micrognathozoa, and Rotifera. Although they do not possess a jaw apparatus, Acanthocephala also belong to Gnathifera due to their epidermal structure. ©Encyclopedia of Life Support Systems (EOLSS) BIOLOGICAL SCIENCE FUNDAMENTALS AND SYSTEMATICS – Vol. -

Phylogeny Inference Based on Parsimony and Other Methods Using Paup*
8 Phylogeny inference based on parsimony and other methods using Paup* THEORY David L. Swofford and Jack Sullivan 8.1 Introduction Methods for inferring evolutionary trees can be divided into two broad categories: thosethatoperateonamatrixofdiscretecharactersthatassignsoneormore attributes or character states to each taxon (i.e. sequence or gene-family member); and those that operate on a matrix of pairwise distances between taxa, with each distance representing an estimate of the amount of divergence between two taxa since they last shared a common ancestor (see Chapter 1). The most commonly employed discrete-character methods used in molecular phylogenetics are parsi- mony and maximum likelihood methods. For molecular data, the character-state matrix is typically an aligned set of DNA or protein sequences, in which the states are the nucleotides A, C, G, and T (i.e. DNA sequences) or symbols representing the 20 common amino acids (i.e. protein sequences); however, other forms of discrete data such as restriction-site presence/absence and gene-order information also may be used. Parsimony, maximum likelihood, and some distance methods are examples of a broader class of phylogenetic methods that rely on the use of optimality criteria. Methods in this class all operate by explicitly defining an objective function that returns a score for any input tree topology. This tree score thus allows any two or more trees to be ranked according to the chosen optimality criterion. Ordinarily, phylogenetic inference under criterion-based methods couples the selection of The Phylogenetic Handbook: a Practical Approach to Phylogenetic Analysis and Hypothesis Testing, Philippe Lemey, Marco Salemi, and Anne-Mieke Vandamme (eds.). -

Number of Living Species in Australia and the World
Numbers of Living Species in Australia and the World 2nd edition Arthur D. Chapman Australian Biodiversity Information Services australia’s nature Toowoomba, Australia there is more still to be discovered… Report for the Australian Biological Resources Study Canberra, Australia September 2009 CONTENTS Foreword 1 Insecta (insects) 23 Plants 43 Viruses 59 Arachnida Magnoliophyta (flowering plants) 43 Protoctista (mainly Introduction 2 (spiders, scorpions, etc) 26 Gymnosperms (Coniferophyta, Protozoa—others included Executive Summary 6 Pycnogonida (sea spiders) 28 Cycadophyta, Gnetophyta under fungi, algae, Myriapoda and Ginkgophyta) 45 Chromista, etc) 60 Detailed discussion by Group 12 (millipedes, centipedes) 29 Ferns and Allies 46 Chordates 13 Acknowledgements 63 Crustacea (crabs, lobsters, etc) 31 Bryophyta Mammalia (mammals) 13 Onychophora (velvet worms) 32 (mosses, liverworts, hornworts) 47 References 66 Aves (birds) 14 Hexapoda (proturans, springtails) 33 Plant Algae (including green Reptilia (reptiles) 15 Mollusca (molluscs, shellfish) 34 algae, red algae, glaucophytes) 49 Amphibia (frogs, etc) 16 Annelida (segmented worms) 35 Fungi 51 Pisces (fishes including Nematoda Fungi (excluding taxa Chondrichthyes and (nematodes, roundworms) 36 treated under Chromista Osteichthyes) 17 and Protoctista) 51 Acanthocephala Agnatha (hagfish, (thorny-headed worms) 37 Lichen-forming fungi 53 lampreys, slime eels) 18 Platyhelminthes (flat worms) 38 Others 54 Cephalochordata (lancelets) 19 Cnidaria (jellyfish, Prokaryota (Bacteria Tunicata or Urochordata sea anenomes, corals) 39 [Monera] of previous report) 54 (sea squirts, doliolids, salps) 20 Porifera (sponges) 40 Cyanophyta (Cyanobacteria) 55 Invertebrates 21 Other Invertebrates 41 Chromista (including some Hemichordata (hemichordates) 21 species previously included Echinodermata (starfish, under either algae or fungi) 56 sea cucumbers, etc) 22 FOREWORD In Australia and around the world, biodiversity is under huge Harnessing core science and knowledge bases, like and growing pressure. -

Going Deeper Into High and Low Phylogenetic Relationships of Protura
G C A T T A C G G C A T genes Article Going Deeper into High and Low Phylogenetic Relationships of Protura 1, , 2,3, 3 1 1 Antonio Carapelli * y , Yun Bu y, Wan-Jun Chen , Francesco Nardi , Chiara Leo , Francesco Frati 1 and Yun-Xia Luan 3,4,* 1 Department of Life Sciences, University of Siena, Via A. Moro 2, 53100 Siena, Italy; [email protected] (F.N.); [email protected] (C.L.); [email protected] (F.F.) 2 Natural History Research Center, Shanghai Natural History Museum, Shanghai Science & Technology Museum, Shanghai 200041, China; [email protected] 3 Key Laboratory of Insect Developmental and Evolutionary Biology, Institute of Plant Physiology and Ecology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai 200032, China; [email protected] 4 Guangdong Provincial Key Laboratory of Insect Developmental Biology and Applied Technology, Institute of Insect Science and Technology, School of Life Sciences, South China Normal University, Guangzhou 510631, China * Correspondence: [email protected] (A.C.); [email protected] (Y.-X.L.); Tel.: +39-0577-234410 (A.C.); +86-18918100826 (Y.-X.L.) These authors contributed equally to this work. y Received: 16 March 2019; Accepted: 5 April 2019; Published: 10 April 2019 Abstract: Proturans are small, wingless, soil-dwelling arthropods, generally associated with the early diversification of Hexapoda. Their bizarre morphology, together with conflicting results of molecular studies, has nevertheless made their classification ambiguous. Furthermore, their limited dispersal capability (due to the primarily absence of wings) and their euedaphic lifestyle have greatly complicated species-level identification. -

Phylogenomic Resolution of Sea Spider Diversification Through Integration Of
bioRxiv preprint doi: https://doi.org/10.1101/2020.01.31.929612; this version posted February 2, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Phylogenomic resolution of sea spider diversification through integration of multiple data classes 1Jesús A. Ballesteros†, 1Emily V.W. Setton†, 1Carlos E. Santibáñez López†, 2Claudia P. Arango, 3Georg Brenneis, 4Saskia Brix, 5Esperanza Cano-Sánchez, 6Merai Dandouch, 6Geoffrey F. Dilly, 7Marc P. Eleaume, 1Guilherme Gainett, 8Cyril Gallut, 6Sean McAtee, 6Lauren McIntyre, 9Amy L. Moran, 6Randy Moran, 5Pablo J. López-González, 10Gerhard Scholtz, 6Clay Williamson, 11H. Arthur Woods, 12Ward C. Wheeler, 1Prashant P. Sharma* 1 Department of Integrative Biology, University of Wisconsin–Madison, Madison, WI, USA 2 Queensland Museum, Biodiversity Program, Brisbane, Australia 3 Zoologisches Institut und Museum, Cytologie und Evolutionsbiologie, Universität Greifswald, Greifswald, Germany 4 Senckenberg am Meer, German Centre for Marine Biodiversity Research (DZMB), c/o Biocenter Grindel (CeNak), Martin-Luther-King-Platz 3, Hamburg, Germany 5 Biodiversidad y Ecología Acuática, Departamento de Zoología, Facultad de Biología, Universidad de Sevilla, Sevilla, Spain 6 Department of Biology, California State University-Channel Islands, Camarillo, CA, USA 7 Départment Milieux et Peuplements Aquatiques, Muséum national d’Histoire naturelle, Paris, France 8 Institut de Systématique, Emvolution, Biodiversité (ISYEB), Sorbonne Université, CNRS, Concarneau, France 9 Department of Biology, University of Hawai’i at Mānoa, Honolulu, HI, USA Page 1 of 31 bioRxiv preprint doi: https://doi.org/10.1101/2020.01.31.929612; this version posted February 2, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. -

Evolution of Pycnogonid Life History Traits Eric Carl Lovely University of New Hampshire, Durham
University of New Hampshire University of New Hampshire Scholars' Repository Doctoral Dissertations Student Scholarship Winter 1999 Evolution of pycnogonid life history traits Eric Carl Lovely University of New Hampshire, Durham Follow this and additional works at: https://scholars.unh.edu/dissertation Recommended Citation Lovely, Eric Carl, "Evolution of pycnogonid life history traits" (1999). Doctoral Dissertations. 1975. https://scholars.unh.edu/dissertation/1975 This Dissertation is brought to you for free and open access by the Student Scholarship at University of New Hampshire Scholars' Repository. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of University of New Hampshire Scholars' Repository. For more information, please contact [email protected]. INFORMATION TO USERS This manuscript has been reproduced from the microfilm master. UMI films the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter face, while others may be from any type of computer printer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely affect reproduction. In the unlikely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. Oversize materials (e.g., maps, drawings, charts) are reproduced by sectioning the original, beginning at the upper left-hand comer and continuing from left to right in equal sections with small overlaps. -

Constraints on the Timescale of Animal Evolutionary History
Palaeontologia Electronica palaeo-electronica.org Constraints on the timescale of animal evolutionary history Michael J. Benton, Philip C.J. Donoghue, Robert J. Asher, Matt Friedman, Thomas J. Near, and Jakob Vinther ABSTRACT Dating the tree of life is a core endeavor in evolutionary biology. Rates of evolution are fundamental to nearly every evolutionary model and process. Rates need dates. There is much debate on the most appropriate and reasonable ways in which to date the tree of life, and recent work has highlighted some confusions and complexities that can be avoided. Whether phylogenetic trees are dated after they have been estab- lished, or as part of the process of tree finding, practitioners need to know which cali- brations to use. We emphasize the importance of identifying crown (not stem) fossils, levels of confidence in their attribution to the crown, current chronostratigraphic preci- sion, the primacy of the host geological formation and asymmetric confidence intervals. Here we present calibrations for 88 key nodes across the phylogeny of animals, rang- ing from the root of Metazoa to the last common ancestor of Homo sapiens. Close attention to detail is constantly required: for example, the classic bird-mammal date (base of crown Amniota) has often been given as 310-315 Ma; the 2014 international time scale indicates a minimum age of 318 Ma. Michael J. Benton. School of Earth Sciences, University of Bristol, Bristol, BS8 1RJ, U.K. [email protected] Philip C.J. Donoghue. School of Earth Sciences, University of Bristol, Bristol, BS8 1RJ, U.K. [email protected] Robert J. -

Chilopoda; Scolopendridae
Bijdragen tot dl Dierkunde, 55 (1): 125-130 — 1985 Possible species isolation mechanisms in some scolopendrid centipedes (Chilopoda; Scolopendridae) by J.G.E. Lewis Taunton School, Taunton, Somerset TA2 6AD, England the femur of Abstract are seen on Eupolybothrus spp. Various other lithobiid secondary sexual sexual characters in dis- Secondary centipedes are briefly characters have been reviewed by Lewis (1981). cussed and it is that the the suggested spines on prefemora of the described Differences type above are of the last pair of legs in some scolopendrids are used in presumably associated with mating behaviour specific discrimination prior to mating. The hypothesis is in which male and female head discussed with reference of the come together to Scolopendra spp. eastern Mediterranean,north-east Africa and Arabia. to tail, and the antennae and the last pair of legs Where species of Scolopendra with identical The virtually are tapped. morphological adaptations in spinulation on the last legs are sympatric, a large size dif- the males would serve for the femalesto identify ference exists between them. different and sex. It is that in where the species suggested some genera prefemoral have been inhibited. In spines are absent, speciation may the where be absent genus Otostigmus spines may or spine similar in number of other patterns are very a species DIMORPHISM IN SCOLOPENDRIDAE secondary sexual characters have developed. Sexual is also in dimorphism seen the scolopen- INTRODUCTION dromorph family Scolopendridae. In Scolopendra morsitans Linnaeus the dorsal side of the Centipedes not infrequently exhibit secondary prefemur, femur and sometimes the tibiaof the last of is sexual characters. -

South Carolina Department of Natural Resources
FOREWORD Abundant fish and wildlife, unbroken coastal vistas, miles of scenic rivers, swamps and mountains open to exploration, and well-tended forests and fields…these resources enhance the quality of life that makes South Carolina a place people want to call home. We know our state’s natural resources are a primary reason that individuals and businesses choose to locate here. They are drawn to the high quality natural resources that South Carolinians love and appreciate. The quality of our state’s natural resources is no accident. It is the result of hard work and sound stewardship on the part of many citizens and agencies. The 20th century brought many changes to South Carolina; some of these changes had devastating results to the land. However, people rose to the challenge of restoring our resources. Over the past several decades, deer, wood duck and wild turkey populations have been restored, striped bass populations have recovered, the bald eagle has returned and more than half a million acres of wildlife habitat has been conserved. We in South Carolina are particularly proud of our accomplishments as we prepare to celebrate, in 2006, the 100th anniversary of game and fish law enforcement and management by the state of South Carolina. Since its inception, the South Carolina Department of Natural Resources (SCDNR) has undergone several reorganizations and name changes; however, more has changed in this state than the department’s name. According to the US Census Bureau, the South Carolina’s population has almost doubled since 1950 and the majority of our citizens now live in urban areas. -
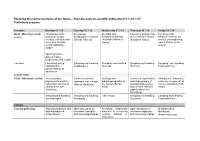
Exploring the Marine Meiofauna of the Azores – from Discovery to Scientific Publication (15.7.-24.7.19) Preliminary Program
Exploring the marine meiofauna of the Azores – from discovery to scientific publication (15.7.-24.7.19) Preliminary program: Schedule Monday 15.7.19 Tuesday 16.7.19 Wednesday 17.7.19 Thursday 18.7.19 Friday 19.7.19 09.00 - Morning session Introduction to the Meiofaunal Intertidal and Taxonomy and diversity The Proseriata Lecture workshop (people, Scalidophora (Andreas meiofaunal annelids of meiofaunal molluscs (Platyhelminthes): the facilities, schedule and Schmidt-Rhaesa) (Alejandro Martínez (Katharina Jörger) interest of unappealing aims; Ana Ricardo Garcia) worms (Marco Curini- Costa/ Katharina Galetti) Jörger) Opening lecture: Azores marine biodiversity (Ana Costa) Lab work Laboratory set-up, Sampling and handling Sampling and handling Sampling and handling Sampling and handling handling and Scalidophora Annelida Mollusca Platyhelminthes documentation of specimens LUNCH TIME 14.00 - Afternoon session Acoelomorpha – Marine nematodes: Geology and Taxonomy, systematics Rhabdocoel flatworms, phylogenetic position, taxonomy and ecology paleobiogeography of and biogeography of a diverse component of systematic and how to (Alberto Navarrete) the Azores (Sérgio meiofaunal Nemertea marine meiofauna (Tom identify them (Ulf Avila) and relevant research Artois) Jondelius) opportunities (Jon Norenburg) Sampling and handling Sampling and handling Lab session Sampling and handling Sampling and handling Acoelomorpha Nematoda Nemertea Platyhelminthes DINNER Evening gathering Sampling strategies and Open discussion on Adressing biodiversity Open discussion