Immunotherapy of Glioblastoma: Current Strategies and Challenges in Tumor Model Development
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Challenge Jogging Province De Liège 2016 8E 15 KM De Liège - 1 Mei 2016 2,7 KM
Challenge Jogging Province de Liège 2016 8e 15 KM de Liège - 1 mei 2016 2,7 KM PL NR Naam Woonplaats Club Age CAT P/CAT Tijd GEM KM-Tijd 1 20301 BARÉ François LIÈGE RFCL ATHLÉTISME 14 esh 1 0:09:08 17,74 3:23 2 46493 ARNONE RFCL 16 esh 2 0:09:10 17,67 3:24 3 20272 SENTERRE Guillaume STE-VÉ 3 SEC 3K 15 esh 3 0:09:26 17,17 3:30 4 20244 MAES Abel STE-VÉ 2 SEC 3K 14 esh 4 0:09:41 16,73 3:35 5 20035 DETHIER Alexandre 11 esh 5 0:09:45 16,62 3:37 6 20045 JAMME Martin LIÈGE RFCL 18 seh 1 0:09:52 16,42 3:39 7 20117 KALIN Lionel HENRI-CHAPELLE HAUTES FAGNES 11 esh 6 0:09:56 16,31 3:41 8 20271 SAMPSON Yanni STE-VÉ 6 PRIM 3 12 esh 7 0:10:01 16,17 3:43 9 20334 VANGEEBERGEN Hugo LIEGE RFCL 12 esh 8 0:10:09 15,96 3:46 10 20228 LAMARCHE Matéo STE-VÉ 1 SEC 3K 13 esh 9 0:10:15 15,80 3:48 11 20221 JADOUL Nicolas STE-VÉ 2 SEC 3K 14 esh 10 0:10:26 15,53 3:52 12 20258 OLIVIER Augustin STE-VÉ 2 SEC 3K 14 esh 11 0:10:32 15,38 3:54 13 20121 PIEDBOEUF Maxime EMBOURG CSM 6 esh 12 0:10:34 15,33 3:55 14 20036 GOULARD Charles Antoine LIÈGE HERSTAL ATHLÉTI 13 esh 13 0:10:35 15,31 3:55 15 20166 DALNE Victor STE-VÉ 2 SEC 3K 14 esh 14 0:10:37 15,26 3:56 16 20262 POLLERS Tim STE-VÉ 2 SEC 3K 14 esh 15 0:10:48 15,00 4:00 17 20180 D'HOEGELEER Pierre STE-VÉ SYMPATHI 14 esh 16 0:10:52 14,91 4:01 18 20288 WILSON Robert STE-VÉ PARENT 3 42 seh 2 0:10:53 14,89 4:02 19 20647 PIERRE Arthur 12 esh 17 0:10:54 14,86 4:02 20 20033 SWINNEN Eliott 8 esh 18 0:10:57 14,79 4:03 21 20154 BUSI Carla STE-VÉ 2 SEC 3K 14 esf 1 0:11:00 14,73 4:04 22 20155 BUSI Sacha STE-VÉ 2 SEC 3K 14 esf 2 0:11:00 -
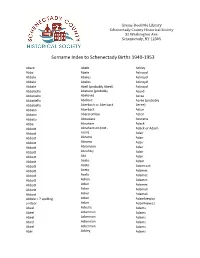
Surname Index to Schenectady Births 1940-1953
Grems-Doolittle Library Schenectady County Historical Society 32 Washington Ave. Schenectady, NY 12305 Surname Index to Schenectady Births 1940-1953 Abare Abele Ackley Abba Abele Ackroyd Abbale Abeles Ackroyd Abbale Abeles Ackroyd Abbale Abell (probably Abeel) Ackroyd Abbatiello Abelone (probably Acord Abbatiello Abelove) Acree Abbatiello Abelove Acree (probably Abbatiello Aberbach or Aberback Aeree) Abbato Aberback Acton Abbato Abercrombie Acton Abbato Aboudara Acucena Abbe Abraham Adack Abbott Abrahamson (not - Adack or Adach Abbott nson) Adair Abbott Abrams Adair Abbott Abrams Adair Abbott Abramson Adair Abbott Abrofsky Adair Abbott Abt Adair Abbott Aceto Adam Abbott Aceto Adamczak Abbott Aceto Adamec Abbott Aceto Adamec Abbott Acken Adamec Abbott Acker Adamec Abbott Acker Adamek Abbott Acker Adamek Abbzle = ? spelling Acker Adamkiewicz unclear Acker Adamkiewicz Abeel Ackerle Adams Abeel Ackerman Adams Abeel Ackerman Adams Abeel Ackerman Adams Abeel Ackerman Adams Abel Ackley Adams Grems-Doolittle Library Schenectady County Historical Society 32 Washington Ave. Schenectady, NY 12305 Surname Index to Schenectady Births 1940-1953 Adams Adamson Ahl Adams Adanti Ahles Adams Addis Ahman Adams Ademec or Adamec Ahnert Adams Adinolfi Ahren Adams Adinolfi Ahren Adams Adinolfi Ahrendtsen Adams Adinolfi Ahrendtsen Adams Adkins Ahrens Adams Adkins Ahrens Adams Adriance Ahrens Adams Adsit Aiken Adams Aeree Aiken Adams Aernecke Ailes = ? Adams Agans Ainsworth Adams Agans Aker (or Aeher = ?) Adams Aganz (Agans ?) Akers Adams Agare or Abare = ? Akerson Adams Agat Akin Adams Agat Akins Adams Agen Akins Adams Aggen Akland Adams Aggen Albanese Adams Aggen Alberding Adams Aggen Albert Adams Agnew Albert Adams Agnew Albert or Alberti Adams Agnew Alberti Adams Agostara Alberti Adams Agostara (not Agostra) Alberts Adamski Agree Albig Adamski Ahave ? = totally Albig Adamson unclear Albohm Adamson Ahern Albohm Adamson Ahl Albohm (not Albolm) Adamson Ahl Albrezzi Grems-Doolittle Library Schenectady County Historical Society 32 Washington Ave. -

ICON S Conference 17 – 19 June 2016 Humboldt University Berlin
Berlin Berlin 2016 ICON S CONFERENCE 17 – 19 JUNE 2016 HUMBOLDT UNIVERSITY BERLIN We extend a warm welcome to all participants in the 2016 Annual Meeting of ICON S, the International Society of Public Law. This will be our largest Annual Meeting since the foundation of the Society in 2013. The panels, roundtables and plenary events address the Conference’s overarching theme of “Borders, Otherness and Public Law” and other topics at the heart of contemporary public law inquiry. We are grateful to our Berlin hosts for their relentless hard work and creativity in putting together such a mega-sized event, and we thank our sponsors for their generous support. Most of all, we thank you, the ICON S members, for your overwhelmingly positive response to the call for papers this year, and for volunteering your time and energy to promote the success of the Society and its annual conference. Together, we have created what we believe is a first-rate, intellectually appealing program featuring scholars, jurists and policy makers from various disciplines and from literally four corners of the world. We hope that you enjoy it thoroughly! Gráinne de Búrca ( New York University ) and Ran Hirschl ( University of Toronto ) Co-Presidents, ICON S, the International Society of Public Law S Conference ICON WElcomE statEMEnts 1 I WELcoME STATEMENTS 1 II ICON S INAUGURAL GOVERNANCE 3 III SCHEDULE 4 IV MAP OF CONFERENCE VENUES 6 V PLENARY EVENTS 7 VI CONCURRING PANELS 15 a OVERVIEW 16 b PANEls SEssion I 26 c PANEls SEssion II 43 D PANEls SEssion III 77 E PANEls SEssion IV 110 F PANEls SEssion V 143 VII ICON S WP CONFERENCE ProcEEDINGS SERIES 179 VIII SERVICE 180 IX PARTICIPANTS 190 ICON S CONFERENCE 17 – 19 JUNE 2016 HUMBOLDT UNIVERSITY BERLIN We extend a warm welcome to all participants in the 2016 Annual Meeting of ICON S, the International Society of Public Law. -

Index to St. Louis, Missouri Naturalization Records Created After Sept
Index to St. Louis, Missouri Naturalization Records Created after Sept. 27, 1906 Alphabetical surname index L–M History & Genealogy Department St. Louis County Library 1640 S. Lindberg Blvd. St. Louis, Missouri 63131 314-994-3300, ext. 2070 [email protected] Index to St. Louis, Missouri Naturalization Records Created after Sept. 27, 1906 This index covers St. Louis, Missouri naturalization records created between October 1, 1906 and December 1928 and is based on the following sources: • Naturalizations, U.S. District Court—Eastern Division, Eastern Judicial District of Missouri, Vols. 1 – 82 • Naturalizations, U.S. Circuit Court— Eastern Division, Eastern Judicial District of Missouri, Vols. 5 – 21 Entries are listed alphabetically by surname, then by given name, and then numerically by volume number. Abbreviations and Notations SLCL = History and Genealogy Department microfilm number (St. Louis County Library) FHL = Family History Library microfilm number * = spelling taken from the signature which differed from name in index. How to obtain copies Photocopies of indexed articles may be requested by sending an email to the History and Genealogy Department at [email protected]. A limit of three searches per request applies. Please review the library's lookup policy at https://www.slcl.org/genealogy-and-local- history/services. A declaration of intention may lead to further records. For more information, contact the National Archives at the address below. Include all information listed on the declaration of intention. National Archives, Central Plains Region 400 W. Pershing Rd. Kansas City, MO 64108 (816) 268-8000 [email protected] History Genealogy Dept. Index to St. Louis, Missouri Naturalization Records St. -
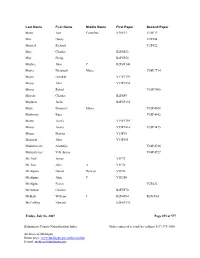
Kalamazoo County Naturalization Index Order Copies of Records by Calling (517) 373-1408
Last Name First Name Middle Name First Paper Second Paper Maus Joos Cornelius V30P33 V30P33 Max Henry V2P204 Maxted Richard V2P422 May Charles B2F6P23 May Philip B2F6P55 Maybee John C B2F6P140 Mayer Elizabeth Marie V58P3714 Mayer Hendrik V17P3359 Mayer John V17P3234 Mayer Robert V51P3086 Mayers Charles B2F6P9 Mayhew Jacob B2F6P135 Mayr Emanuele Maria V62P4308 Mazkrists Ruta V65P4942 Mazur Josefa V19P3789 Mazur Josefa V55P3416 V55P3415 Mazur Marcin V15P18 Mazurak John V13P291 Mazurkevics Anatolijs V64P4726 Mazurkevics Vilhelmina V64P4727 Mc Neil James V5P72 Mc Rae John A V5P76 McAlpine Daniel Duncan V3P26 McAlpine John C V3P250 McAlpine Peeter V2P426 McArthur Charles B2F5P70 McBeth William L B2F6P84 B2F6P84 McCaffrey Edward B2F6P136 Friday, July 06, 2007 Page 299 of 577 Kalamazoo County Naturalization Index Order copies of records by calling (517) 373-1408 Archives of Michigan Home page: www.michigan.gov/archivesofmi E-mail: [email protected] Last Name First Name Middle Name First Paper Second Paper McCall Hannah V16P3084 McCallum Duncan Clyde V10P370 McCallum Duncan Clyke V23P212 V23P212 McCally Geoffrey Thomas V52P3165 McCamby Alex B2F6P1 McCamly Daniel B2F6P5 McCandless Frances Georgina Forsythe V61P4262 McCandless Hugh Milligan V20P4331 McCandry Daniel B2F6P5 McClelland George Charles V23P101 McClure Mary Abernethy V10P389 McClurky Charles B2F6P67 McCormick George W V5P33 McCormick Olga V48P2744 McCray Robert B2F6P157 McCudden John B2F6P150 McCue Hugh B2F5P56 McCue John B2F6P76 McCue Peter V62P4379 McCugan Vincent Murray V19P3955 -

Supplemental Appendix S1. COMPASS Outcome Definitions
Supplemental Appendix S1. COMPASS outcome definitions TABLE OF CONTENTS 1.0 MYOCARDIAL INFARCTION 3 1.1 Non-procedural MI 3 1.2 Peri-procedural MI 3 1.21 PCI-related MI 3 1.22 CABG, transcatheter aortic valve or mitral clip -related MI 3 1.23 MI related to other cardiac procedures 3 1.24 MI associated with non-cardiac procedures (within 48 hours of procedure) 3 1.3 Probable MI 4 2.0 ANGINA 4 2.1 Unstable angina 4 2.2 Worsening angina 4 2.3 New angina 4 3.0 STENT THROMBOSIS 4 3.1 Definite stent thrombosis 4 3.2 Probable stent thrombosis 5 4.0 CORONARY ARTERY BYPASS GRAFT FAILURE 5 5.0 HEART FAILURE (NEW OR WORSENING) 5 6.0 STROKE 5 6.1 Definite ischemic stroke 6 6.2 Definite hemorrhagic stroke 6 6.21 Primary intracerebral / intraparenchymal / intraventricular brain hemorrhage 6 6.22 Subarachnoid hemorrhage 6 6.3 Uncertain or unknown stroke 6 7.0 TRANSIENT ISCHEMIC ATTACK 6 8.0 DEATH 6 8.1 Non-cardiovascular death 6 8.11 Malignancy death 6 8.12 Other non-cardiovascular death not due to malignancy or bleeding 7 8.13 Fatal bleeding other than due to hemorrhagic stroke 7 8.2 Cardiovascular death 7 8.21 CV death within 30 days of acute MI 7 8.22 CV death within 30 days of Stroke 7 8.23 CV death within 14 days of Heart failure 7 8.24 CV Death within 3 days of a CV procedure 7 8.25 Sudden cardiac death 7 8.26 Death due to other cardiovascular cause 8 8.27 Death due to unknown cause 8 9.0 RESUSCITATED CARDIAC ARREST 8 10.0 SEVERE/ACUTE LIMB ISCHEMIA, PERIPHERAL VASCULAR INTERVENTION AND PERIPHERAL VASCULAR HOSPITALIZATION 8 10.1 Acute limb ischemia -

Classic 24 Hour of Daytona - Group A
Classic 24 Hour of Daytona - Group A Team Name Car Info Car Number Driver's Name (First, Last) Driver Country Dean DeSantis US D&L Vintage 67 Alfa Romeo GT Jr 5 Kenneth Moore US Klub Sport 67 Porsche 911S 71 George Calfo US Marc Devis Belgium Sasco Sports 65 Lola T70 Mk1 11 Christian Traber Switzerland R.E. Farrell UK R.E. Farrell 62 Jaguar E Type 17 Robert Gate UK Christian Dumolin Belgium GipiMotors 65 Ford Mustang 21 Christophe Van Riet Belgium Chris Wilson Nigel Williams Australia Lanzante 65 Ford GT40 MK1 27 Dean Lanzante UK Peter Mulder Germany 69 Porsche 911 S 28 Patrick Simon Germany Grahame Bryant UK Type 1 Performance 67 Chevrolet Camaro 29 Oliver Bryant UK Philip Walker UK Philip Walker 65 Ford GT40 37 Mike Jordan UK Alan Terpins Brazil 68 Porsche 911 T/R 99 Andre Lara Resende US 70 Porsche 914-6 GT 40 Jon Wactor US Gray Gregory US J&L Fabricating 70 Chevron B16 44 Randy Buck US Chris Woodgate UK Mark Midgley UK Sean Kukula UK Rex J Woodgate 64 Elva Courier MK4 49 Martin Greaves UK Alan Benjamin CO Patrick Long CA Alan Benjamin 67 Porsche 911S 54 Hurley Haywood FL GipiMotors 70 Lola T70 MkIIIb 70 Thierry De Latre Du Bosqueau Belgium BGM Sport 70 Ferrari Daytona 365 GT 77 Tim Summers UK Jurgen Barth Germany Willi Kauhsen Racing 67 Porsche 907 Long Tail Mauro Casadai Italy 66 Ginetta G4 Leigh Davis UK 66 Porsche Carrera Mario Sala Italy Cherry Howard US Grant Motorsports 66 Porsche 910 39 John Higgins US Heritage Motorsports 69 BMW 2002 Tii 147 Russell Gee US Ford Fairlane Markku Biederman Germany Craig Watkins MJE Motorsports 68 Porsche 911 11 Johannes Van Overbeek Lee Chapman Racing Lola T70 T.B.D. -

Shorter Versus Longer Durations of Exclusive Human Milk Feeding and Diabetes Outcomes in Offspring: a Systematic Review
United States Department of Agriculture Shorter Versus Longer Durations of Exclusive Human Milk Feeding and Diabetes Outcomes in Offspring: A Systematic Review The Pregnancy and Birth to 24 Months Project Published date: April 15, 2019 Nutrition Evidence Systematic Review Center for Nutrition Policy and Promotion Food and Nutrition Service U.S. Department of Agriculture 3101 Park Center Drive Alexandria, Virginia This systematic review was conducted for the Pregnancy and Birth to 24 Months Project (P/B-24 Project) by the Nutrition Evidence Systematic Review (NESR) team at the Center for Nutrition Policy and Promotion, Food and Nutrition Service, USDA. All systematic reviews from the P/B-24 Project are available on the NESR website: https://nesr.usda.gov. Conclusion statements drawn as part of this systematic review describe the state of science related to the specific question examined. Conclusion statements do not draw implications, and should not be interpreted as dietary guidance. The contents of this document may be used and reprinted without permission. Endorsements by NESR, the Center for Nutrition Policy and Promotion, the Food and Nutrition Service, or the U.S. Department of Agriculture (USDA) of derivative products developed from this work may not be stated or implied. In accordance with Federal civil rights law and USDA civil rights regulations and policies, the USDA, its Agencies, offices, and employees, and institutions participating in or administering USDA programs are prohibited from discriminating based on race, color, national origin, religion, sex, gender identity (including gender expression), sexual orientation, disability, age, marital status, family/parental status, income derived from a public assistance program, political beliefs, or reprisal or retaliation for prior civil rights activity, in any program or activity conducted or funded by USDA (not all bases apply to all programs). -

Ridgewater College Deans' List – Spring Semester 2021
RIDGEWATER COLLEGE DEANS’ LIST – SPRING SEMESTER 2021 Campus Full Name Hometown Campus Full Name Hometown Willmar Connor Aalderks Sacred Heart Hutchinson Caleb Bonick Waconia Willmar Hillary Adams Kandiyohi Willmar Chelsey Borg Hutchinson Willmar Jamie Adams Little Falls Hutchinson Charlene Botones Winsted Willmar Dennis Aguilar Willmar Willmar Morgan Boucher Cold Spring Willmar Harlee Ahrens Redwood Falls Hutchinson Christine Bowles Howard Lake Hutchinson Jessica Albrecht Stewart Hutchinson David Brandon Hutchinson Hutchinson Remington Albright Waverly Willmar Makayla Breth Albany Hutchinson Ryce Alexander Waconia Hutchinson Mateo Briseno Dassel Willmar Abdijabar Ali Willmar Willmar Kaitlin Bruns Morris Willmar Elizabeth Amelung Sauk Centre Hutchinson Steven Brustuen Grove City Hutchinson Brenna Amundsen Dassel Hutchinson Michael Brutger Annandale Hutchinson Kurt Andersen St Michael Willmar Tyler Bulthuis Spicer Willmar Alyssa Anderson Lewisville Willmar Desiree Burggraff Redfield, SD Willmar Austen Anderson Willmar Hutchinson Charles Busche Dassel Willmar Brad Anderson Sunburg Willmar Omar Bustos Willmar Willmar Emma Anderson Cambridge Willmar Amanda Bzdok St Cloud Willmar Karissa Anderson Willmar Hutchinson James Calhoon Litchfield Willmar Kassidy Anderson Watertown, SD Hutchinson Juan Cambara Echeverria Monticello Willmar Kellie Anderson Ashby Hutchinson Christopher Campbell New Hope Willmar Nicole Anderson Willmar Hutchinson Jose Campos Hutchinson Willmar Cheyenne Appleman Bath, PA Hutchinson Brian Cannon Litchfield Willmar Seth -

Toxic and Teratogenic Effects of Prenatal Alcohol Exposure on Fetal Development, Adolescence, and Adulthood
International Journal of Molecular Sciences Review Toxic and Teratogenic Effects of Prenatal Alcohol Exposure on Fetal Development, Adolescence, and Adulthood Dae D. Chung , Marisa R. Pinson, Lokeshwar S. Bhenderu , Michael S. Lai, Rhea A. Patel and Rajesh C. Miranda * Department of Neuroscience and Experimental Therapeutics, Texas A&M University Health Science Center, Texas A&M University, Bryan, TX 77807, USA; [email protected] (D.D.C.); [email protected] (M.R.P.); [email protected] (L.S.B.); [email protected] (M.S.L.); [email protected] (R.A.P.) * Correspondence: [email protected]; Tel.: +1-979-436-0332; Fax: +1-979-436-0086 Abstract: Prenatal alcohol exposure (PAE) can have immediate and long-lasting toxic and teratogenic effects on an individual’s development and health. As a toxicant, alcohol can lead to a variety of physical and neurological anomalies in the fetus that can lead to behavioral and other impairments which may last a lifetime. Recent studies have focused on identifying mechanisms that mediate the immediate teratogenic effects of alcohol on fetal development and mechanisms that facilitate the persistent toxic effects of alcohol on health and predisposition to disease later in life. This review focuses on the contribution of epigenetic modifications and intercellular transporters like extracellular vesicles to the toxicity of PAE and to immediate and long-term consequences on an individual’s health and risk of disease. Citation: Chung, D.D.; Pinson, M.R.; Keywords: prenatal alcohol exposure; alcohol; early childhood adversity; extracellular vesicles; Bhenderu, L.S.; Lai, M.S.; Patel, R.A.; developmental origin of health and disease; epigenetic modification; miRNA Miranda, R.C. -

Deans' List | Spring 2021
COLLEGE of EDUCATION and HUMAN SCIENCES DEANS’ LIST | SPRING 2021 A Emma Barnes Caughlin Bohn Percy Caraig Katrina Barnes Kennedy Bork Alyssa Carlson Abdulaali Abdulaali Hayden Barrows Alyssa Borson Amy Carlson Eva Achtenberg Baylie Bass Hailey Bottoms Anna Carlson Makala Acker Carlie Bates Jessica Bousquet Jenna Carlson Allison Aden Mya Battaglia Kasey Brabec Robin Carlson Maddie Aerts Lexi Bay Mackenzie Brabec Cayley Carpenter Fatima Al-jibory Emma Baynes Jerratt Bradley Samantha Carr Sarah Al-Tewaig Kierra Bearinger Sam Bradney Grace Carter Jenny Aldana Allie Beberniss Paige Brandenburg Ma’Kiya Carter Monica Aldrighetti Allyson Beck Jayven Brandt Kaitlin Cartwright Tori Allan McKenna Beezley Jordan Brandt Cooper Caskey Theodore Allen Daisy Beltran Alejandra Brekke-Diaz Carly Castle Kiley Allgood Myah Beltran Michael Brick Jadyn Cattau Evan Anderson Caroline Bendig Tanner Brickman Madison Cave Rachel Anderson Brooke Bennett Kaidence Brodersen Jalea Chandler Skylar Anderson Jessica Bennett Kyla Bronson Cade Chapin Claire Andry Logan Bennett Jadyn Brooks Brooklyn Childears Cody Anschutz Sidney Benson Paris Brooks Halle Childers Meghan Arena Sydney Benson Brenna Brown Payton Chmelka Maya Arrigo Tiera Berggren Matt Broyles Colby Chohrach Emma Arthur Teryn Berks Carly Brune Carly Christensen Audrey Aschoff Alexa Berney Cassidee Bruntz Ella Christensen Breanna Ashton Isabelle Bertrand Kamryn Buchanan Maizie Christensen Libby Atherton Alison Best Alexis Buchert Sydney Christensen Jackson Atwell Melissa Bevins Jessica Buckles Dylan Christian -

Government Gazette Staatskoerant REPUBLIC of SOUTH AFRICA REPUBLIEK VAN SUID-AFRIKA
Government Gazette Staatskoerant REPUBLIC OF SOUTH AFRICA REPUBLIEK VAN SUID-AFRIKA July Vol. 625 Pretoria, 7 2017 Julie No. 40961 PART 1 OF 2 LEGAL NOTICES A WETLIKE KENNISGEWINGS ISSN 1682-5843 N.B. The Government Printing Works will 40961 not be held responsible for the quality of “Hard Copies” or “Electronic Files” submitted for publication purposes 9 771682 584003 AIDS HELPLINE: 0800-0123-22 Prevention is the cure 2 No. 40961 GOVERNMENT GAZETTE, 7 JULY 2017 IMPORTANT NOTICE: THE GOVERNMENT PRINTING WORKS WILL NOT BE HELD RESPONSIBLE FOR ANY ERRORS THAT MIGHT OCCUR DUE TO THE SUBMISSION OF INCOMPLETE / INCORRECT / ILLEGIBLE COPY. NO FUTURE QUERIES WILL BE HANDLED IN CONNECTION WITH THE ABOVE. Table of Contents LEGAL NOTICES BUSINESS NOTICES • BESIGHEIDSKENNISGEWINGS Gauteng ....................................................................................................................................... 12 Eastern Cape / Oos-Kaap ................................................................................................................. 13 Western Cape / Wes-Kaap ................................................................................................................ 13 COMPANY NOTICES • MAATSKAPPYKENNISGEWINGS Gauteng ....................................................................................................................................... 13 Eastern Cape / Oos-Kaap ................................................................................................................. 14 KwaZulu-Natal ...............................................................................................................................