Phospholipase D Functional Ablation Has a Protective Effect in An
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplemental Figure 1. Vimentin
Double mutant specific genes Transcript gene_assignment Gene Symbol RefSeq FDR Fold- FDR Fold- FDR Fold- ID (single vs. Change (double Change (double Change wt) (single vs. wt) (double vs. single) (double vs. wt) vs. wt) vs. single) 10485013 BC085239 // 1110051M20Rik // RIKEN cDNA 1110051M20 gene // 2 E1 // 228356 /// NM 1110051M20Ri BC085239 0.164013 -1.38517 0.0345128 -2.24228 0.154535 -1.61877 k 10358717 NM_197990 // 1700025G04Rik // RIKEN cDNA 1700025G04 gene // 1 G2 // 69399 /// BC 1700025G04Rik NM_197990 0.142593 -1.37878 0.0212926 -3.13385 0.093068 -2.27291 10358713 NM_197990 // 1700025G04Rik // RIKEN cDNA 1700025G04 gene // 1 G2 // 69399 1700025G04Rik NM_197990 0.0655213 -1.71563 0.0222468 -2.32498 0.166843 -1.35517 10481312 NM_027283 // 1700026L06Rik // RIKEN cDNA 1700026L06 gene // 2 A3 // 69987 /// EN 1700026L06Rik NM_027283 0.0503754 -1.46385 0.0140999 -2.19537 0.0825609 -1.49972 10351465 BC150846 // 1700084C01Rik // RIKEN cDNA 1700084C01 gene // 1 H3 // 78465 /// NM_ 1700084C01Rik BC150846 0.107391 -1.5916 0.0385418 -2.05801 0.295457 -1.29305 10569654 AK007416 // 1810010D01Rik // RIKEN cDNA 1810010D01 gene // 7 F5 // 381935 /// XR 1810010D01Rik AK007416 0.145576 1.69432 0.0476957 2.51662 0.288571 1.48533 10508883 NM_001083916 // 1810019J16Rik // RIKEN cDNA 1810019J16 gene // 4 D2.3 // 69073 / 1810019J16Rik NM_001083916 0.0533206 1.57139 0.0145433 2.56417 0.0836674 1.63179 10585282 ENSMUST00000050829 // 2010007H06Rik // RIKEN cDNA 2010007H06 gene // --- // 6984 2010007H06Rik ENSMUST00000050829 0.129914 -1.71998 0.0434862 -2.51672 -

Role of Phospholipases in Adrenal Steroidogenesis
229 1 W B BOLLAG Phospholipases in adrenal 229:1 R29–R41 Review steroidogenesis Role of phospholipases in adrenal steroidogenesis Wendy B Bollag Correspondence should be addressed Charlie Norwood VA Medical Center, One Freedom Way, Augusta, GA, USA to W B Bollag Department of Physiology, Medical College of Georgia, Augusta University (formerly Georgia Regents Email University), Augusta, GA, USA [email protected] Abstract Phospholipases are lipid-metabolizing enzymes that hydrolyze phospholipids. In some Key Words cases, their activity results in remodeling of lipids and/or allows the synthesis of other f adrenal cortex lipids. In other cases, however, and of interest to the topic of adrenal steroidogenesis, f angiotensin phospholipases produce second messengers that modify the function of a cell. In this f intracellular signaling review, the enzymatic reactions, products, and effectors of three phospholipases, f phospholipids phospholipase C, phospholipase D, and phospholipase A2, are discussed. Although f signal transduction much data have been obtained concerning the role of phospholipases C and D in regulating adrenal steroid hormone production, there are still many gaps in our knowledge. Furthermore, little is known about the involvement of phospholipase A2, Endocrinology perhaps, in part, because this enzyme comprises a large family of related enzymes of that are differentially regulated and with different functions. This review presents the evidence supporting the role of each of these phospholipases in steroidogenesis in the Journal Journal of Endocrinology adrenal cortex. (2016) 229, R1–R13 Introduction associated GTP-binding protein exchanges a bound GDP for a GTP. The G protein with GTP bound can then Phospholipids serve a structural function in the cell in that activate the enzyme, phospholipase C (PLC), that cleaves they form the lipid bilayer that maintains cell integrity. -

Phospholipase D in Cell Proliferation and Cancer
Vol. 1, 789–800, September 2003 Molecular Cancer Research 789 Subject Review Phospholipase D in Cell Proliferation and Cancer David A. Foster and Lizhong Xu The Department of Biological Sciences, Hunter College of The City University of New York, New York, NY Abstract trafficking, cytoskeletal reorganization, receptor endocytosis, Phospholipase D (PLD) has emerged as a regulator of exocytosis, and cell migration (4, 5). A role for PLD in cell several critical aspects of cell physiology. PLD, which proliferation is indicated from reports showing that PLD catalyzes the hydrolysis of phosphatidylcholine (PC) to activity is elevated in response to platelet-derived growth factor phosphatidic acid (PA) and choline, is activated in (PDGF; 6), fibroblast growth factor (7, 8), epidermal growth response to stimulators of vesicle transport, endocyto- factor (EGF; 9), insulin (10), insulin-like growth factor 1 (11), sis, exocytosis, cell migration, and mitosis. Dysregula- growth hormone (12), and sphingosine 1-phosphate (13). PLD tion of these cell biological processes occurs in the activity is also elevated in cells transformed by a variety development of a variety of human tumors. It has now of transforming oncogenes including v-Src (14), v-Ras (15, 16), been observed that there are abnormalities in PLD v-Fps (17), and v-Raf (18). Thus, there is a growing body of expression and activity in many human cancers. In this evidence linking PLD activity with mitogenic signaling. While review, evidence is summarized implicating PLD as a PLD has been associated with many aspects of cell physiology critical regulator of cell proliferation, survival signaling, such as membrane trafficking and cytoskeletal organization cell transformation, and tumor progression. -

Phospholipase D in Cell Proliferation and Cancer
Vol. 1, 789–800, September 2003 Molecular Cancer Research 789 Subject Review Phospholipase D in Cell Proliferation and Cancer David A. Foster and Lizhong Xu The Department of Biological Sciences, Hunter College of The City University of New York, New York, NY Abstract trafficking, cytoskeletal reorganization, receptor endocytosis, Phospholipase D (PLD) has emerged as a regulator of exocytosis, and cell migration (4, 5). A role for PLD in cell several critical aspects of cell physiology. PLD, which proliferation is indicated from reports showing that PLD catalyzes the hydrolysis of phosphatidylcholine (PC) to activity is elevated in response to platelet-derived growth factor phosphatidic acid (PA) and choline, is activated in (PDGF; 6), fibroblast growth factor (7, 8), epidermal growth response to stimulators of vesicle transport, endocyto- factor (EGF; 9), insulin (10), insulin-like growth factor 1 (11), sis, exocytosis, cell migration, and mitosis. Dysregula- growth hormone (12), and sphingosine 1-phosphate (13). PLD tion of these cell biological processes occurs in the activity is also elevated in cells transformed by a variety development of a variety of human tumors. It has now of transforming oncogenes including v-Src (14), v-Ras (15, 16), been observed that there are abnormalities in PLD v-Fps (17), and v-Raf (18). Thus, there is a growing body of expression and activity in many human cancers. In this evidence linking PLD activity with mitogenic signaling. While review, evidence is summarized implicating PLD as a PLD has been associated with many aspects of cell physiology critical regulator of cell proliferation, survival signaling, such as membrane trafficking and cytoskeletal organization cell transformation, and tumor progression. -

Expression Profiling of Palmitate
Expression Profiling of Palmitate- and Oleate-Regulated Genes Provides Novel Insights Into the Effects of Chronic Lipid Exposure on Pancreatic -Cell Function Anna K. Busch,1 Damien Cordery,1 Gareth S. Denyer,2 and Trevor J. Biden1 Chronic lipid exposure is implicated in -cell dysfunc- tion in type 2 diabetes. We therefore used oligonucleo- tide arrays to define global alterations in gene irculating nutrients regulate the function of pan- expression in MIN6 cells after 48-h pretreatment with creatic -cells at multiple levels: they acutely oleate or palmitate. Altogether, 126 genes were altered stimulate insulin secretion, modulate insulin >1.9-fold by palmitate, 62 by oleate, and 46 by both biosynthesis, and, over the longer term, bring lipids. Importantly, nine of the palmitate-regulated C about adaptive changes in gene expression (1). Dysregu- genes are known to be correspondingly changed in  lation of any of these aspects of the nutrient response models of type 2 diabetes. A tendency toward -cell  de-differentiation was also apparent with palmitate: and/or alterations in -cell differentiation and survival are pyruvate carboxylase and mitochondrial glycerol potentially implicated in the onset of type 2 diabetes. This 3-phosphate dehydrogenase were downregulated, disease is associated with peripheral insulin resistance but whereas lactate dehydrogenase and fructose 1,6- only develops in conjunction with a failure of -cell bisphosphatases were induced. Increases in the latter compensation, which otherwise counteracts the insulin (also seen with oleate), along with glucosamine-phos- resistance (2–5). Type 2 diabetic patients display reduc- phate N-acetyl transferase, imply upregulation of the tions in -cell mass as compared with insulin-resistant hexosamine biosynthesis pathway in palmitate-treated cells. -

Interface of Phospholipase Activity, Immune Cell Function, and Atherosclerosis
biomolecules Review Interface of Phospholipase Activity, Immune Cell Function, and Atherosclerosis Robert M. Schilke y, Cassidy M. R. Blackburn y , Temitayo T. Bamgbose and Matthew D. Woolard * Department of Microbiology and Immunology, Louisiana State University Health Sciences Center, Shreveport, LA 71130, USA; [email protected] (R.M.S.); [email protected] (C.M.R.B.); [email protected] (T.T.B.) * Correspondence: [email protected]; Tel.: +1-(318)-675-4160 These authors contributed equally to this work. y Received: 12 September 2020; Accepted: 13 October 2020; Published: 15 October 2020 Abstract: Phospholipases are a family of lipid-altering enzymes that can either reduce or increase bioactive lipid levels. Bioactive lipids elicit signaling responses, activate transcription factors, promote G-coupled-protein activity, and modulate membrane fluidity, which mediates cellular function. Phospholipases and the bioactive lipids they produce are important regulators of immune cell activity, dictating both pro-inflammatory and pro-resolving activity. During atherosclerosis, pro-inflammatory and pro-resolving activities govern atherosclerosis progression and regression, respectively. This review will look at the interface of phospholipase activity, immune cell function, and atherosclerosis. Keywords: atherosclerosis; phospholipases; macrophages; T cells; lipins 1. Introduction All cellular membranes are composed mostly of phospholipids. Phospholipids are amphiphilic compounds with a hydrophilic, negatively charged phosphate group head and two hydrophobic fatty acid tail residues [1]. The glycerophospholipids, phospholipids with glycerol backbones, are the largest group of phospholipids, which are classified by the modification of the head group [1]. The negatively charged phosphate head forms an ionic bond with an amino alcohol. This bridges the glycerol backbone to the nitrogenous functional group (amino alcohol). -

A Role for Phospholipase D1 in Neurotransmitter Release
A role for phospholipase D1 in neurotransmitter release Yann Humeau*, Nicolas Vitale*, Sylvette Chasserot-Golaz*, Jean-Luc Dupont*, Guangwei Du†, Michael A. Frohman†, Marie-France Bader*, and Bernard Poulain*‡ *Centre National de la Recherche Scientifique, Unite´Propre de Recherche 2356, Neurotransmission et Se´cre´tion Neuroendocrine, 5 Rue Blaise Pascal, IFR37, 67084 Strasbourg, France; and †Department of Pharmacology and Center for Developmental Genetics, University Medical Center, Stony Brook, NY 11794-5140 Edited by Erwin Neher, Max Planck Institute for Biophysical Chemistry, Goettingen, Germany, and approved October 23, 2001 (received for review July 13, 2001) Phosphatidic acid produced by phospholipase D (PLD) as a result of PLD1 plays an important role in Ca2ϩ-dependent exocytosis, signaling activity is thought to play a role in membrane vesicle operating a late stage subsequent to the cytoskeletal-mediated trafficking, either as an intracellular messenger or as a cone-shaped recruitment of secretory granules to exocytotic sites (14). We lipid that promotes membrane fusion. We recently described that, proposed that PLD1 constitutes a key factor for the late fusion in neuroendocrine cells, plasma membrane-associated PLD1 oper- event. Because Ca2ϩ-dependent membrane fusion proceeds ؉ ates at a stage of Ca2 -dependent exocytosis subsequent to cy- through a closely related mechanism in secretory cells and toskeletal-mediated recruitment of secretory granules to exocy- neurons, our results raised the question of whether PLD1 also totic sites. We show here that PLD1 also plays a crucial role in may play a role in neurotransmitter release. We show here that neurotransmitter release. Using purified rat brain synaptosomes PLD1 is present in rat nerve terminals at the presynaptic plasma subjected to hypotonic lysis and centrifugation, we found that membrane. -

Supplemental Figures 04 12 2017
Jung et al. 1 SUPPLEMENTAL FIGURES 2 3 Supplemental Figure 1. Clinical relevance of natural product methyltransferases (NPMTs) in brain disorders. (A) 4 Table summarizing characteristics of 11 NPMTs using data derived from the TCGA GBM and Rembrandt datasets for 5 relative expression levels and survival. In addition, published studies of the 11 NPMTs are summarized. (B) The 1 Jung et al. 6 expression levels of 10 NPMTs in glioblastoma versus non‐tumor brain are displayed in a heatmap, ranked by 7 significance and expression levels. *, p<0.05; **, p<0.01; ***, p<0.001. 8 2 Jung et al. 9 10 Supplemental Figure 2. Anatomical distribution of methyltransferase and metabolic signatures within 11 glioblastomas. The Ivy GAP dataset was downloaded and interrogated by histological structure for NNMT, NAMPT, 12 DNMT mRNA expression and selected gene expression signatures. The results are displayed on a heatmap. The 13 sample size of each histological region as indicated on the figure. 14 3 Jung et al. 15 16 Supplemental Figure 3. Altered expression of nicotinamide and nicotinate metabolism‐related enzymes in 17 glioblastoma. (A) Heatmap (fold change of expression) of whole 25 enzymes in the KEGG nicotinate and 18 nicotinamide metabolism gene set were analyzed in indicated glioblastoma expression datasets with Oncomine. 4 Jung et al. 19 Color bar intensity indicates percentile of fold change in glioblastoma relative to normal brain. (B) Nicotinamide and 20 nicotinate and methionine salvage pathways are displayed with the relative expression levels in glioblastoma 21 specimens in the TCGA GBM dataset indicated. 22 5 Jung et al. 23 24 Supplementary Figure 4. -
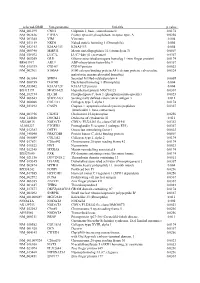
Supplementary Figures S1-S3
selected-GBID Uni-genename Uni-title p value NM_001299 CNN1 Calponin 1, basic, smooth muscle 0.0174 NM_002836 PTPRA Protein tyrosine phosphatase, receptor type, A 0.0256 NM_003380 VIM Vimentin 0.004 NM_033119 NKD1 Naked cuticle homolog 1 (Drosophila) 0.004 NM_052913 KIAA1913 KIAA1913 0.004 NM_005940 MMP11 Matrix metallopeptidase 11 (stromelysin 3) 0.0069 NM_018032 LUC7L LUC7-like (S. cerevisiae) 0.0367 NM_005269 GLI1 Glioma-associated oncogene homolog 1 (zinc finger protein) 0.0174 BE463997 ARL9 ADP-ribosylation factor-like 9 0.0367 NM_015939 CGI-09 CGI-09 protein 0.0023 NM_002961 S100A4 S100 calcium binding protein A4 (calcium protein, calvasculin, 0.0324 metastasin, murine placental homolog) NM_003014 SFRP4 Secreted frizzled-related protein 4 0.0005 NM_080759 DACH1 Dachshund homolog 1 (Drosophila) 0.004 NM_053042 KIAA1729 KIAA1729 protein 0.004 BX415194 MGC16121 Hypothetical protein MGC16121 0.0367 NM_182734 PLCB1 Phospholipase C, beta 1 (phosphoinositide-specific) 0.0023 NM_006643 SDCCAG3 Serologically defined colon cancer antigen 3 0.011 NM_000088 COL1A1 Collagen, type I, alpha 1 0.0174 NM_033292 CASP1 Caspase 1, apoptosis-related cysteine peptidase 0.0367 (interleukin 1, beta, convertase) NM_003956 CH25H Cholesterol 25-hydroxylase 0.0256 NM_144658 DOCK11 Dedicator of cytokinesis 11 0.011 AK024935 NODATA CDNA: FLJ21283 fis, clone COL01910 0.0363 AL050227 PTGER3 Prostaglandin E receptor 3 (subtype EP3) 0.0367 NM_012383 OSTF1 Osteoclast stimulating factor 1 0.0023 NM_145040 PRKCDBP Protein kinase C, delta binding protein 0.0069 NM_000089 -

Dynamic Phospholipid Signaling by G Protein-Coupled Receptors ⁎ Paschal A
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Biochimica et Biophysica Acta 1768 (2007) 888–900 www.elsevier.com/locate/bbamem Review Dynamic phospholipid signaling by G protein-coupled receptors ⁎ Paschal A. Oude Weernink a, , Li Han a, Karl H. Jakobs a, Martina Schmidt b a Institut für Pharmakologie, Universitätsklinikum Essen, Hufelandstrasse 55, 45122 Essen, Germany b Department of Molecular Pharmacology, University of Groningen, 9713 AV Groningen, The Netherlands Received 28 June 2006; received in revised form 18 September 2006; accepted 18 September 2006 Available online 23 September 2006 Abstract G protein-coupled receptors (GPCRs) control a variety of fundamental cellular processes by regulating phospholipid signaling pathways. Essential for signaling by a large number of receptors is the hydrolysis of the membrane phosphoinositide PIP2 by phospholipase C (PLC) into the second messengers IP3 and DAG. Many receptors also stimulate phospholipase D (PLD), leading to the generation of the versatile lipid, phosphatidic acid. Particular PLC and PLD isoforms take differential positions in receptor signaling and are additionally regulated by small GTPases of the Ras, Rho and ARF families. It is now recognized that the PLC substrate, PIP2, has signaling capacity by itself and can, by direct interaction, affect the activity and subcellular localization of PLD and several other proteins. As expected, the synthesis of PIP2 by phosphoinositide 5-kinases is tightly regulated as well. In this review, we present an overview of how these signaling pathways are governed by GPCRs, explain the molecular basis for the spatially and temporally organized, highly dynamic quality of phospholipid signaling, and point to the functional connection of the pathways. -

Downloaded from the Mouse Lysosome Gene Database, Mlgdb
1 Supplemental Figure Legends 2 3 Supplemental Figure S1: Epidermal-specific mTORC1 gain-of-function models show 4 increased mTORC1 activation and down-regulate EGFR and HER2 protein expression in a 5 mTORC1-sensitive manner. (A) Immunoblotting of Rheb1 S16H flox/flox keratinocyte cultures 6 infected with empty or adenoviral cre recombinase for markers of mTORC1 (p-S6, p-4E-BP1) 7 activity. (B) Tsc1 cKO epidermal lysates also show decreased expression of TSC2 by 8 immunoblotting of the same experiment as in Figure 2A. (C) Immunoblotting of Tsc2 flox/flox 9 keratinocyte cultures infected with empty or adenoviral cre recombinase showing decreased EGFR 10 and HER2 protein expression. (D) Expression of EGFR and HER2 was decreased in Tsc1 cre 11 keratinocytes compared to empty controls, and up-regulated in response to Torin1 (1µM, 24 hrs), 12 by immunoblot analyses. Immunoblots are contemporaneous and parallel from the same biological 13 replicate and represent the same experiment as depicted in Figure 7B. (E) Densitometry 14 quantification of representative immunoblot experiments shown in Figures 2E and S1D (r≥3; error 15 bars represent STDEV; p-values by Student’s T-test). 16 17 18 19 20 21 22 23 Supplemental Figure S2: EGFR and HER2 transcription are unchanged with epidermal/ 24 keratinocyte Tsc1 or Rptor loss. Egfr and Her2 mRNA levels in (A) Tsc1 cKO epidermal lysates, 25 (B) Tsc1 cKO keratinocyte lysates and(C) Tsc1 cre keratinocyte lysates are minimally altered 26 compared to their respective controls. (r≥3; error bars represent STDEV; p-values by Student’s T- 27 test). -

A Feedback Mechanism Between Phospholipase D and Deadenylase PARN for the Shortening of Eukaryotic Poly(A) Mrna Tails That Is Deregulated in Cancer Cells
A Feedback Mechanism between Phospholipase D and Deadenylase PARN for the Shortening of Eukaryotic Poly(A) mRNA Tails that is Deregulated in Cancer Cells Taylor E. Miller2 and Julian Gomez-Cambronero1,2* 1Center for Experimental Therapeutics and Reperfusion Injury, Brigham and Women’s Hospital and Harvard Medical School, Boston, MA 02115. 2Wright State University School of Medicine, Department of Biochemistry and Molecular Biology, Dayton, Ohio 45435. *Corresponding author. Address: Wright State University Boonshoft School of Medicine, Department of Biochemistry and Molecular Biology, 3640 Colonel Glenn Highway, Dayton, OH 45435, USA. Phone: 937-775-4767. Fax: 937-775-3730. E-mail: [email protected]. © 2016. Published by The Company of Biologists Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed. Biology Open • Advance article Abbreviations: BSA, bovine serum albumin; DMEM, Dulbecco’s Modified Eagle Media; DOPA, 1,2-dioleoyl-sn-glycero-3-phosphate; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; FCS, fetal calf serum; FITC, fluorescein isothiocyanate; mTOR, mammalian target of rapamycin; PA, phosphatidic acid; PARN, poly(A) specific- ribonuclease; PBS, phosphate buffered saline; PC, phosphatidylcholine; PLD, phospholipase D; PH, phox homology domain; PLD2, phospholipase D2; PP2A, phosphoprotein 2A; PVDF, polyvinylidene difluoride; RNase, ribonuclease; TRITC, tetramethylrhodamine isothiocyanate; PAN, RNA polyadenylated nuclear complex. Biology Open • Advance article ABSTRACT The removal of mRNA transcripts poly(A) tails by 3-5 exonucleases is the rate-limiting step in mRNA decay in eukaryotes.