Ribosomopathies: How a Common Root Can Cause a Tree of Pathologies Nadia Danilova1,* and Hanna T
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Data
Figure 2S 4 7 A - C 080125 CSCs 080418 CSCs - + IFN-a 48 h + IFN-a 48 h + IFN-a 72 h 6 + IFN-a 72 h 3 5 MRFI 4 2 3 2 1 1 0 0 MHC I MHC II MICA MICB ULBP-1 ULBP-2 ULBP-3 ULBP-4 MHC I MHC II MICA MICB ULBP-1 ULBP-2 ULBP-3 ULBP-4 7 B 13 080125 FBS - D 080418 FBS - + IFN-a 48 h 12 + IFN-a 48 h + IFN-a 72 h + IFN-a 72 h 6 080125 FBS 11 10 5 9 8 4 7 6 3 MRFI 5 4 2 3 2 1 1 0 0 MHC I MHC II MICA MICB ULBP-1 ULBP-2 ULBP-3 ULBP-4 MHC I MHC II MICA MICB ULBP-1 ULBP-2 ULBP-3 ULBP-4 Molecule Molecule FIGURE 4S FIGURE 5S Panel A Panel B FIGURE 6S A B C D Supplemental Results Table 1S. Modulation by IFN-α of APM in GBM CSC and FBS tumor cell lines. Molecule * Cell line IFN-α‡ HLA β2-m# HLA LMP TAP1 TAP2 class II A A HC§ 2 7 10 080125 CSCs - 1∞ (1) 3 (65) 2 (91) 1 (2) 6 (47) 2 (61) 1 (3) 1 (2) 1 (3) + 2 (81) 11 (80) 13 (99) 1 (3) 8 (88) 4 (91) 1 (2) 1 (3) 2 (68) 080125 FBS - 2 (81) 4 (63) 4 (83) 1 (3) 6 (80) 3 (67) 2 (86) 1 (3) 2 (75) + 2 (99) 14 (90) 7 (97) 5 (75) 7 (100) 6 (98) 2 (90) 1 (4) 3 (87) 080418 CSCs - 2 (51) 1 (1) 1 (3) 2 (47) 2 (83) 2 (54) 1 (4) 1 (2) 1 (3) + 2 (81) 3 (76) 5 (75) 2 (50) 2 (83) 3 (71) 1 (3) 2 (87) 1 (2) 080418 FBS - 1 (3) 3 (70) 2 (88) 1 (4) 3 (87) 2 (76) 1 (3) 1 (3) 1 (2) + 2 (78) 7 (98) 5 (99) 2 (94) 5 (100) 3 (100) 1 (4) 2 (100) 1 (2) 070104 CSCs - 1 (2) 1 (3) 1 (3) 2 (78) 1 (3) 1 (2) 1 (3) 1 (3) 1 (2) + 2 (98) 8 (100) 10 (88) 4 (89) 3 (98) 3 (94) 1 (4) 2 (86) 2 (79) * expression of APM molecules was evaluated by intracellular staining and cytofluorimetric analysis; ‡ cells were treatead or not (+/-) for 72 h with 1000 IU/ml of IFN-α; # β-2 microglobulin; § β-2 microglobulin-free HLA-A heavy chain; ∞ values are indicated as ratio between the mean of fluorescence intensity of cells stained with the selected mAb and that of the negative control; bold values indicate significant MRFI (≥ 2). -

Supplementary Materials: Evaluation of Cytotoxicity and Α-Glucosidase Inhibitory Activity of Amide and Polyamino-Derivatives of Lupane Triterpenoids
Supplementary Materials: Evaluation of cytotoxicity and α-glucosidase inhibitory activity of amide and polyamino-derivatives of lupane triterpenoids Oxana B. Kazakova1*, Gul'nara V. Giniyatullina1, Akhat G. Mustafin1, Denis A. Babkov2, Elena V. Sokolova2, Alexander A. Spasov2* 1Ufa Institute of Chemistry of the Ufa Federal Research Centre of the Russian Academy of Sciences, 71, pr. Oktyabrya, 450054 Ufa, Russian Federation 2Scientific Center for Innovative Drugs, Volgograd State Medical University, Novorossiyskaya st. 39, Volgograd 400087, Russian Federation Correspondence Prof. Dr. Oxana B. Kazakova Ufa Institute of Chemistry of the Ufa Federal Research Centre of the Russian Academy of Sciences 71 Prospeсt Oktyabrya Ufa, 450054 Russian Federation E-mail: [email protected] Prof. Dr. Alexander A. Spasov Scientific Center for Innovative Drugs of the Volgograd State Medical University 39 Novorossiyskaya st. Volgograd, 400087 Russian Federation E-mail: [email protected] Figure S1. 1H and 13C of compound 2. H NH N H O H O H 2 2 Figure S2. 1H and 13C of compound 4. NH2 O H O H CH3 O O H H3C O H 4 3 Figure S3. Anticancer screening data of compound 2 at single dose assay 4 Figure S4. Anticancer screening data of compound 7 at single dose assay 5 Figure S5. Anticancer screening data of compound 8 at single dose assay 6 Figure S6. Anticancer screening data of compound 9 at single dose assay 7 Figure S7. Anticancer screening data of compound 12 at single dose assay 8 Figure S8. Anticancer screening data of compound 13 at single dose assay 9 Figure S9. Anticancer screening data of compound 14 at single dose assay 10 Figure S10. -

Allele-Specific Expression of Ribosomal Protein Genes in Interspecific Hybrid Catfish
Allele-specific Expression of Ribosomal Protein Genes in Interspecific Hybrid Catfish by Ailu Chen A dissertation submitted to the Graduate Faculty of Auburn University in partial fulfillment of the requirements for the Degree of Doctor of Philosophy Auburn, Alabama August 1, 2015 Keywords: catfish, interspecific hybrids, allele-specific expression, ribosomal protein Copyright 2015 by Ailu Chen Approved by Zhanjiang Liu, Chair, Professor, School of Fisheries, Aquaculture and Aquatic Sciences Nannan Liu, Professor, Entomology and Plant Pathology Eric Peatman, Associate Professor, School of Fisheries, Aquaculture and Aquatic Sciences Aaron M. Rashotte, Associate Professor, Biological Sciences Abstract Interspecific hybridization results in a vast reservoir of allelic variations, which may potentially contribute to phenotypical enhancement in the hybrids. Whether the allelic variations are related to the downstream phenotypic differences of interspecific hybrid is still an open question. The recently developed genome-wide allele-specific approaches that harness high- throughput sequencing technology allow direct quantification of allelic variations and gene expression patterns. In this work, I investigated allele-specific expression (ASE) pattern using RNA-Seq datasets generated from interspecific catfish hybrids. The objective of the study is to determine the ASE genes and pathways in which they are involved. Specifically, my study investigated ASE-SNPs, ASE-genes, parent-of-origins of ASE allele and how ASE would possibly contribute to heterosis. My data showed that ASE was operating in the interspecific catfish system. Of the 66,251 and 177,841 SNPs identified from the datasets of the liver and gill, 5,420 (8.2%) and 13,390 (7.5%) SNPs were identified as significant ASE-SNPs, respectively. -

The Oviductal Extracellular Vesicles' RNA Cargo Regulates the Bovine
International Journal of Molecular Sciences Article The Oviductal Extracellular Vesicles’ RNA Cargo Regulates the Bovine Embryonic Transcriptome Stefan Bauersachs 1 , Pascal Mermillod 2 and Carmen Almiñana 1,2,* 1 Genetics and Functional Genomics, VetSuisse Faculty Zurich, University of Zurich, 8315 Lindau (ZH), Switzerland; [email protected] 2 UMR85 PRC, INRA, CNRS 7247, Université de Tours, IFCE, 37380 Nouzilly, France; [email protected] * Correspondence: [email protected] Received: 21 January 2020; Accepted: 12 February 2020; Published: 14 February 2020 Abstract: Oviductal extracellular vesicles (oEVs) are emerging as key players in the gamete/embryo–oviduct interactions that contribute to successful pregnancy. Various positive effects of oEVs on gametes and early embryos have been found in vitro. To determine whether these effects are associated with changes of embryonic gene expression, the transcriptomes of embryos supplemented with bovine fresh (FeEVs) or frozen (FoEVs) oEVs during in vitro culture compared to controls without oEVs were analyzed by low-input RNA sequencing. Analysis of RNA-seq data revealed 221 differentially expressed genes (DEGs) between FoEV treatment and control, 67 DEGs for FeEV and FoEV treatments, and minor differences between FeEV treatment and control (28 DEGs). An integrative analysis of mRNAs and miRNAs contained in oEVs obtained in a previous study with embryonic mRNA alterations pointed to direct effects of oEV cargo on embryos (1) by increasing the concentration of delivered transcripts; (2) by translating delivered mRNAs to proteins that regulate embryonic gene expression; and (3) by oEV-derived miRNAs which downregulate embryonic mRNAs or modify gene expression in other ways. Our study provided the first high-throughput analysis of the embryonic transcriptome regulated by oEVs, increasing our knowledge on the impact of oEVs on the embryo and revealing the oEV RNA components that potentially regulate embryonic development. -

1 AGING Supplementary Table 2
SUPPLEMENTARY TABLES Supplementary Table 1. Details of the eight domain chains of KIAA0101. Serial IDENTITY MAX IN COMP- INTERFACE ID POSITION RESOLUTION EXPERIMENT TYPE number START STOP SCORE IDENTITY LEX WITH CAVITY A 4D2G_D 52 - 69 52 69 100 100 2.65 Å PCNA X-RAY DIFFRACTION √ B 4D2G_E 52 - 69 52 69 100 100 2.65 Å PCNA X-RAY DIFFRACTION √ C 6EHT_D 52 - 71 52 71 100 100 3.2Å PCNA X-RAY DIFFRACTION √ D 6EHT_E 52 - 71 52 71 100 100 3.2Å PCNA X-RAY DIFFRACTION √ E 6GWS_D 41-72 41 72 100 100 3.2Å PCNA X-RAY DIFFRACTION √ F 6GWS_E 41-72 41 72 100 100 2.9Å PCNA X-RAY DIFFRACTION √ G 6GWS_F 41-72 41 72 100 100 2.9Å PCNA X-RAY DIFFRACTION √ H 6IIW_B 2-11 2 11 100 100 1.699Å UHRF1 X-RAY DIFFRACTION √ www.aging-us.com 1 AGING Supplementary Table 2. Significantly enriched gene ontology (GO) annotations (cellular components) of KIAA0101 in lung adenocarcinoma (LinkedOmics). Leading Description FDR Leading Edge Gene EdgeNum RAD51, SPC25, CCNB1, BIRC5, NCAPG, ZWINT, MAD2L1, SKA3, NUF2, BUB1B, CENPA, SKA1, AURKB, NEK2, CENPW, HJURP, NDC80, CDCA5, NCAPH, BUB1, ZWILCH, CENPK, KIF2C, AURKA, CENPN, TOP2A, CENPM, PLK1, ERCC6L, CDT1, CHEK1, SPAG5, CENPH, condensed 66 0 SPC24, NUP37, BLM, CENPE, BUB3, CDK2, FANCD2, CENPO, CENPF, BRCA1, DSN1, chromosome MKI67, NCAPG2, H2AFX, HMGB2, SUV39H1, CBX3, TUBG1, KNTC1, PPP1CC, SMC2, BANF1, NCAPD2, SKA2, NUP107, BRCA2, NUP85, ITGB3BP, SYCE2, TOPBP1, DMC1, SMC4, INCENP. RAD51, OIP5, CDK1, SPC25, CCNB1, BIRC5, NCAPG, ZWINT, MAD2L1, SKA3, NUF2, BUB1B, CENPA, SKA1, AURKB, NEK2, ESCO2, CENPW, HJURP, TTK, NDC80, CDCA5, BUB1, ZWILCH, CENPK, KIF2C, AURKA, DSCC1, CENPN, CDCA8, CENPM, PLK1, MCM6, ERCC6L, CDT1, HELLS, CHEK1, SPAG5, CENPH, PCNA, SPC24, CENPI, NUP37, FEN1, chromosomal 94 0 CENPL, BLM, KIF18A, CENPE, MCM4, BUB3, SUV39H2, MCM2, CDK2, PIF1, DNA2, region CENPO, CENPF, CHEK2, DSN1, H2AFX, MCM7, SUV39H1, MTBP, CBX3, RECQL4, KNTC1, PPP1CC, CENPP, CENPQ, PTGES3, NCAPD2, DYNLL1, SKA2, HAT1, NUP107, MCM5, MCM3, MSH2, BRCA2, NUP85, SSB, ITGB3BP, DMC1, INCENP, THOC3, XPO1, APEX1, XRCC5, KIF22, DCLRE1A, SEH1L, XRCC3, NSMCE2, RAD21. -

Supplementary File 2A Revised
Supplementary file 2A. Differentially expressed genes in aldosteronomas compared to all other samples, ranked according to statistical significance. Missing values were not allowed in aldosteronomas, but to a maximum of five in the other samples. Acc UGCluster Name Symbol log Fold Change P - Value Adj. P-Value B R99527 Hs.8162 Hypothetical protein MGC39372 MGC39372 2,17 6,3E-09 5,1E-05 10,2 AA398335 Hs.10414 Kelch domain containing 8A KLHDC8A 2,26 1,2E-08 5,1E-05 9,56 AA441933 Hs.519075 Leiomodin 1 (smooth muscle) LMOD1 2,33 1,3E-08 5,1E-05 9,54 AA630120 Hs.78781 Vascular endothelial growth factor B VEGFB 1,24 1,1E-07 2,9E-04 7,59 R07846 Data not found 3,71 1,2E-07 2,9E-04 7,49 W92795 Hs.434386 Hypothetical protein LOC201229 LOC201229 1,55 2,0E-07 4,0E-04 7,03 AA454564 Hs.323396 Family with sequence similarity 54, member B FAM54B 1,25 3,0E-07 5,2E-04 6,65 AA775249 Hs.513633 G protein-coupled receptor 56 GPR56 -1,63 4,3E-07 6,4E-04 6,33 AA012822 Hs.713814 Oxysterol bining protein OSBP 1,35 5,3E-07 7,1E-04 6,14 R45592 Hs.655271 Regulating synaptic membrane exocytosis 2 RIMS2 2,51 5,9E-07 7,1E-04 6,04 AA282936 Hs.240 M-phase phosphoprotein 1 MPHOSPH -1,40 8,1E-07 8,9E-04 5,74 N34945 Hs.234898 Acetyl-Coenzyme A carboxylase beta ACACB 0,87 9,7E-07 9,8E-04 5,58 R07322 Hs.464137 Acyl-Coenzyme A oxidase 1, palmitoyl ACOX1 0,82 1,3E-06 1,2E-03 5,35 R77144 Hs.488835 Transmembrane protein 120A TMEM120A 1,55 1,7E-06 1,4E-03 5,07 H68542 Hs.420009 Transcribed locus 1,07 1,7E-06 1,4E-03 5,06 AA410184 Hs.696454 PBX/knotted 1 homeobox 2 PKNOX2 1,78 2,0E-06 -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Micrornas Mediated Regulation of the Ribosomal Proteins and Its Consequences on the Global Translation of Proteins
cells Review microRNAs Mediated Regulation of the Ribosomal Proteins and Its Consequences on the Global Translation of Proteins Abu Musa Md Talimur Reza 1,2 and Yu-Guo Yuan 1,3,* 1 Jiangsu Co-Innovation Center of Prevention and Control of Important Animal Infectious Diseases and Zoonoses, College of Veterinary Medicine, Yangzhou University, Yangzhou 225009, China; [email protected] 2 Institute of Biochemistry and Biophysics, Polish Academy of Sciences, Pawi´nskiego5a, 02-106 Warsaw, Poland 3 Jiangsu Key Laboratory of Zoonosis/Joint International Research Laboratory of Agriculture and Agri-Product Safety, The Ministry of Education of China, Yangzhou University, Yangzhou 225009, China * Correspondence: [email protected]; Tel.: +86-514-8797-9228 Abstract: Ribosomal proteins (RPs) are mostly derived from the energy-consuming enzyme families such as ATP-dependent RNA helicases, AAA-ATPases, GTPases and kinases, and are important structural components of the ribosome, which is a supramolecular ribonucleoprotein complex, composed of Ribosomal RNA (rRNA) and RPs, coordinates the translation and synthesis of proteins with the help of transfer RNA (tRNA) and other factors. Not all RPs are indispensable; in other words, the ribosome could be functional and could continue the translation of proteins instead of lacking in some of the RPs. However, the lack of many RPs could result in severe defects in the biogenesis of ribosomes, which could directly influence the overall translation processes and global expression of the proteins leading to the emergence of different diseases including cancer. While microRNAs (miRNAs) are small non-coding RNAs and one of the potent regulators of the post-transcriptional 0 gene expression, miRNAs regulate gene expression by targeting the 3 untranslated region and/or coding region of the messenger RNAs (mRNAs), and by interacting with the 50 untranslated region, Citation: Reza, A.M.M.T.; Yuan, Y.-G. -

The Role of Human Ribosomal Proteins in the Maturation of Rrna and Ribosome Production
JOBNAME: RNA 14#9 2008 PAGE: 1 OUTPUT: Friday August 8 17:34:50 2008 csh/RNA/164293/rna11320 Downloaded from rnajournal.cshlp.org on September 27, 2021 - Published by Cold Spring Harbor Laboratory Press The role of human ribosomal proteins in the maturation of rRNA and ribosome production SARA ROBLEDO,1,3 RACHEL A. IDOL,1,3 DAN L. CRIMMINS,2 JACK H. LADENSON,2 PHILIP J. MASON,1,4 and MONICA BESSLER1,4 1Department of Internal Medicine, Division of Hematology, Washington University School of Medicine, St. Louis, Missouri 63110, USA 2Department of Pathology and Immunology, Division of Laboratory and Genomic Medicine, Washington University School of Medicine, St. Louis, Missouri 63110, USA ABSTRACT Production of ribosomes is a fundamental process that occurs in all dividing cells. It is a complex process consisting of the coordinated synthesis and assembly of four ribosomal RNAs (rRNA) with about 80 ribosomal proteins (r-proteins) involving more than 150 nonribosomal proteins and other factors. Diamond Blackfan anemia (DBA) is an inherited red cell aplasia caused by mutations in one of several r-proteins. How defects in r-proteins, essential for proliferation in all cells, lead to a human disease with a specific defect in red cell development is unknown. Here, we investigated the role of r-proteins in ribosome biogenesis in order to find out whether those mutated in DBA have any similarities. We depleted HeLa cells using siRNA for several individual r-proteins of the small (RPS6, RPS7, RPS15, RPS16, RPS17, RPS19, RPS24, RPS25, RPS28) or large subunit (RPL5, RPL7, RPL11, RPL14, RPL26, RPL35a) and studied the effect on rRNA processing and ribosome production. -
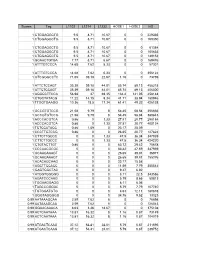
Scores Tag L1102 L1214 L1232 HOSE1 HOSE2 HS 1
Scores Tag L1102 L1214 L1232 HOSE1 HOSE2 HS 1 CTGGAGGCTG 9.5 8.71 10.67 0 0 229335 1 CTGGAGGCTG 9.5 8.71 10.67 0 0 169350 1 CTGGAGGCTG 9.5 8.71 10.67 0 0 61384 1 CTGGAGGCTG 9.5 8.71 10.67 0 0 105633 1 CTGGAGGCTG 9.5 8.71 10.67 0 0 149152 1 GCAACTGTGA 7.77 8.71 6.67 0 0 169476 1 ATTTGTCCCA 14.68 7.62 5.33 0 0 57301 1 ATTTGTCCCA 14.68 7.62 5.33 0 0 356122 1 GTCGGGCCTC 71.65 39.18 22.67 1.16 0 73769 1 ATTCTCCAGT 35.39 39.18 44.01 85.74 89.13 458218 1 ATTCTCCAGT 35.39 39.18 44.01 85.74 89.13 406300 1 AGGGCTTCCA 56.98 37 69.35 134.4 141.05 458148 1 CTGCTATACG 11.22 14.15 9.34 41.71 38.94 180946 1 TTGGTGAAGG 10.36 18.5 17.34 61.41 49.32 426138 1 GCCGTGTCCG 21.58 9.79 8 54.45 58.84 356666 1 GCCGTGTCCG 21.58 9.79 8 54.45 58.84 380843 1 ACCCACGTCA 0.86 0 1.33 27.81 20.77 298184 1 ACCCACGTCA 0.86 0 1.33 27.81 20.77 400124 1 TCTCCATACC 0.86 1.09 0 23.17 25.09 1 CCCTTGTCCG 0.86 0 0 26.65 20.77 127824 1 CTTCTTGCCC 0 0 1.33 47.5 36.34 347939 1 CTTCTTGCCC 0 0 1.33 47.5 36.34 424220 1 CTGTACTTGT 0.86 0 0 63.72 29.42 75678 1 CCCAACGCGC 0 0 0 83.42 47.59 347939 1 GCAAGAAAGT 0 0 0 26.65 39.81 36977 1 GCAAGAAAGT 0 0 0 26.65 39.81 155376 1 ACACAGCAAG 0 0 0 23.17 15.58 1 AGCTTCCACC 0 0 0 11.59 7.79 355542 1 GAGTGGCTAC 0 0 0 9.27 6.92 1 ATGGTGGGGG 0 0 0 8.11 22.5 343586 1 AGATCCCAAG 0 0 0 5.79 8.65 50813 1 TGGAAGGAGG 0 0 0 8.11 6.06 1 TAGCCGGGAC 0 0 0 5.79 7.79 107740 1 TGTGGATGTG 0 0 0 4.63 12.11 180878 1 GGGTAGGGGG 0 0 0 34.76 9.52 13323 0.99 AATAAAGCAA 2.59 7.62 8 0 0 76698 0.99 AATAAAGCAA 2.59 7.62 8 0 0 126043 0.99 GGAACAAACA 8.63 3.26 18.67 0 0 375108 -

RPSA Gene Ribosomal Protein SA
RPSA gene ribosomal protein SA Normal Function The RPSA gene provides instructions for making a protein called ribosomal protein SA, which is one of approximately 80 different ribosomal proteins. These proteins come together to form structures called ribosomes. Ribosomes process the cell's genetic instructions to create proteins. Each ribosome is made up of two parts (subunits) called the large subunit and the small subunit. Ribosomal protein SA is part of the small subunit. The specific roles of each of the ribosomal proteins within the ribosome are not entirely understood. Some ribosomal proteins are involved in the assembly or stability of ribosomes. Others help carry out the ribosome's main function of building new proteins. Research suggests that ribosomal protein SA helps the ribosome control the production of certain proteins, many of which are likely important for development before birth. Health Conditions Related to Genetic Changes Isolated congenital asplenia At least 20 RPSA gene mutations have been identified in individuals with isolated congenital asplenia. People with this condition do not have a spleen but have no other developmental abnormalities. The spleen plays an important role in the immune system. Without this organ, affected individuals are highly susceptible to bacterial infections, which can be life-threatening. RPSA gene mutations are thought to reduce the amount of functional ribosomal protein SA. A shortage of the normal protein likely impairs the assembly of ribosomes, but the specific effects of the mutations -

Ribosomal Proteins Expression and Phylogeny in Alpaca (Lama Pacos) Skin*
Advances in Bioscience and Biotechnology, 2012, 3, 231-237 ABB http://dx.doi.org/10.4236/abb.2012.33032 Published Online June 2012 (http://www.SciRP.org/journal/abb/) Ribosomal proteins expression and phylogeny in alpaca * (Lama pacos) skin Junming Bai, Ruiwen Fan, Shanshan Yang, Yuankai Ji, Jianshan Xie, Chang-Sheng Dong# College of Animal Science and Veterinary Medicine, Shanxi Agricultural University, Taigu, China Email: #[email protected] Received 3 February 2012; revised 8 March 2012; accepted 24 April 2012 ABSTRACT folding/chaperone role in facilitating the processing and folding of rRNA during biogenesis and stabilizing the Ribosomal proteins (RP) has been reported as a cen- mature particle during protein synthesis [3]. It has been tral player in the translation system, and are required well established that global regulation of protein synthe- for the growth and maintenance of all cell kinds. RP sis in eukaryotes is mainly achieved by posttranslational genes form a family of homologous proteins that ex- modification (PTM) of translation factors in response to press in the stable pattern and were used for phy- environmental cues [4]. logenetic analysis. Here we constructed a cDNA li- The family of South American camelids with four brary of alpaca skin and 13,800 clones were sequenced. members is recognized as two wild species, the gua- In the cDNA library, RP genes from skin cDNA li- naco (Lama guanicoe) and the vicuna (Vicugna vicugna) brary of alpaca were identified. Then 8 RP genes were and two of domestic species, the alpaca (Lama pacos) selected at random and built the phylogenetic trees and the llama [5].