KLF13 Loss-Of-Function Variation Contributes to Familial Congenital Heart Defects
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Reversal of Glucocorticoid Resistance in Paediatric Acute Lymphoblastic Leukaemia Is Dependent on Restoring BIM Expression
www.nature.com/bjc ARTICLE Translational Therapeutics Reversal of glucocorticoid resistance in paediatric acute lymphoblastic leukaemia is dependent on restoring BIM expression Cara E. Toscan1, Duohui Jing1, Chelsea Mayoh1 and Richard B. Lock1 BACKGROUND: Acute lymphoblastic leukaemia (ALL) is the most common paediatric malignancy. Glucocorticoids form a critical component of chemotherapy regimens and resistance to glucocorticoid therapy is predictive of poor outcome. We have previously shown that glucocorticoid resistance is associated with upregulation of the oncogene C-MYC and failure to induce the proapoptotic gene BIM. METHODS: A high-throughput screening (HTS) campaign was carried out to identify glucocorticoid sensitisers against an ALL xenograft derived from a glucocorticoid-resistant paediatric patient. Gene expression analysis was carried out using Illumina microarrays. Efficacy, messenger RNA and protein analysis were carried out by Resazurin assay, reverse transcription-PCR and immunoblotting, respectively. RESULTS: A novel glucocorticoid sensitiser, 2-((4,5-dihydro-1H-imidazol-2-yl)thio)-N-isopropyl-N-phenylacetamide (GCS-3), was identified from the HTS campaign. The sensitising effect was specific to glucocorticoids and synergy was observed in a range of dexamethasone-resistant and dexamethasone-sensitive xenografts representative of B-ALL, T-ALL and Philadelphia chromosome- positive ALL. GCS-3 in combination with dexamethasone downregulated C-MYC and significantly upregulated BIM expression in a glucocorticoid-resistant ALL xenograft. The GCS-3/dexamethasone combination significantly increased binding of the glucocorticoid receptor to a novel BIM enhancer, which is associated with glucocorticoid sensitivity. CONCLUSIONS: This study describes the potential of the novel glucocorticoid sensitiser, GCS-3, as a biological tool to interrogate glucocorticoid action and resistance. -

Tgfβ-Regulated Gene Expression by Smads and Sp1/KLF-Like Transcription Factors in Cancer VOLKER ELLENRIEDER
ANTICANCER RESEARCH 28 : 1531-1540 (2008) Review TGFβ-regulated Gene Expression by Smads and Sp1/KLF-like Transcription Factors in Cancer VOLKER ELLENRIEDER Signal Transduction Laboratory, Internal Medicine, Department of Gastroenterology and Endocrinology, University of Marburg, Marburg, Germany Abstract. Transforming growth factor beta (TGF β) controls complex induces the canonical Smad signaling molecules which vital cellular functions through its ability to regulate gene then translocate into the nucleus to regulate transcription (2). The expression. TGFβ binding to its transmembrane receptor cellular response to TGF β can be extremely variable depending kinases initiates distinct intracellular signalling cascades on the cell type and the activation status of a cell at a given time. including the Smad signalling and transcription factors and also For instance, TGF β induces growth arrest and apoptosis in Smad-independent pathways. In normal epithelial cells, TGF β healthy epithelial cells, whereas it can also promote tumor stimulation induces a cytostatic program which includes the progression through stimulation of cell proliferation and the transcriptional repression of the c-Myc oncogene and the later induction of an epithelial-to-mesenchymal transition of tumor induction of the cell cycle inhibitors p15 INK4b and p21 Cip1 . cells (1, 3). In the last decade it has become clear that both the During carcinogenesis, however, many tumor cells lose their tumor suppressing and the tumor promoting functions of TGF β ability to respond to TGF β with growth inhibition, and instead, are primarily regulated on the level of gene expression through activate genes involved in cell proliferation, invasion and Smad-dependent and -independent mechanisms (1, 2, 4). -

Forced Activation of Notch in Macrophages Represses Tumor Growth By
Author Manuscript Published OnlineFirst on January 12, 2016; DOI: 10.1158/0008-5472.CAN-15-2019 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Forced Activation of Notch in Macrophages Represses Tumor Growth by Upregulating miR-125a and Disabling Tumor-Associated Macrophages Jun-Long Zhao1, Fei Huang1, Fei He2, Chun-Chen Gao1, Shi-Qian Liang1, Peng-Fei Ma2, Guang-Ying Dong1, Hua Han1, 2, Hong-Yan Qin1 1State Key Laboratory of Cancer Biology, Department of Medical Genetics and Developmental Biology, 2Department of Hepatic Surgery, Xijing Hospital, Fourth Military Medical University, Xi’an 710032, China Corresponding Authors: Hong-Yan Qin: Department of Medical Genetics and Developmental Biology, Fourth Military Medical University, Chang-Le Xi Street #169, Xi’an 710032, China; Tel.: +86 29 84774487; Fax: +86 29 83246270; Email: [email protected] Hua Han: Department of Medical Genetics and Developmental Biology, Fourth Military Medical University, Chang-Le Xi Street #169, Xi’an 710032, China; Tel.: +86 29 84774513, Fax: +86 29 83246270; Email: [email protected] Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/). J-L. Zhao and F. Huang contributed equally to this study. Running title: Notch regulates TAMs through miR-125a Disclosure of Potential Conflicts of Interest: The authors disclose no potential conflicts of interest. 1 Downloaded from cancerres.aacrjournals.org on September 28, 2021. © 2016 American Association for Cancer Research. Author Manuscript Published OnlineFirst on January 12, 2016; DOI: 10.1158/0008-5472.CAN-15-2019 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. -
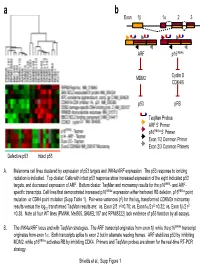
Taqman Probes ARF 5' Primer P16ink4a 5' Primer Exon 1/2
a b Exon 1β 1α 23 ARF p16INK4a MDM2 Cyclin D CDK4/6 p53 pRB TaqMan Probes ARF 5’ Primer p16INK4a 5’ Primer Exon 1/2 Common Primer Exon 2/3 Common Primers Defective p53 Intact p53 A. Melanoma cell lines clustered by expression of p53 targets and INK4a/ARF expression. The p53 response to ionizing radiation is indicated. Top cluster: Cells with intact p53 response show increased expression of the eight indicated p53 targets, and decreased expression of ARF. Bottom cluster: TaqMan and microarray results for the p16INK4- and ARF- specific transcripts. Cell lines that demonstrated increased p16INK4a expression either harbored RB deletion, p16INK4a point 2 mutation or CDK4 point mutation (Supp Table 1). Pair-wise variances (r ) for the log2 transformed CDKN2a microarray 2 2 2 results versus the log10 transformed TaqMan results are: vs. Exon 2/3 r =0.78; vs. Exon1α/2 r =0.32; vs. Exon 1β/2 r =0.38. Note all four WT lines (PMWK, Mel505, SKMEL187 and RPMI8322) lack evidence of p53 function by all assays. B. The INK4a/ARF locus and with TaqMan strategies. The ARF transcript originates from exon 1β while the p16INK4a transcript originates from exon 1α. Both transcripts splice to exon 2 but in alternate reading frames. ARF stabilizes p53 by inhibiting MDM2, while p16INK4a activates RB by inhibiting CDK4. Primers and TaqMan probes are shown for the real-time RT-PCR strategy. Shields et al., Supp Figure 1 SKMEL 28 U01 24h SKMEL WM2664 U01 48h WM2664 U01 24h 24 U01 48h SKMEL 24 U01 24h SKMEL 24 Untreated SKMEL 24 DMSO 48h SKMEL 24 DMSO 24h SKMEL WM2664 DMSO 24h WM2664 DMSO 48h WM2644 Untreated 28 Untreated SKMEL 28 DMSO 48h SKMEL 28 DMSO 24h SKMEL -3.00 -2.00 -1.00 0.00 1.00 2.00 3.00 relative to median expression Genes decreased by UO1 (863) HIF1A Hypoxia-inducible factor 1, alpha subunit NM_001530 RBBP8 Retinoblastoma binding protein 8 NM_002894 Homo sapiens, clone IMAGE:4337652, mRNA BC018676 EIF4EBP1 Eukaryotic translation initiation factor 4E binding protein EXOSC8 Exosome component 8 NM_181503 ENST00000321524 MCM7 MCM7 minichromosome maintenance deficient 7 (S. -

Integrated Computational Approach to the Analysis of RNA-Seq Data Reveals New Transcriptional Regulators of Psoriasis
OPEN Experimental & Molecular Medicine (2016) 48, e268; doi:10.1038/emm.2016.97 & 2016 KSBMB. All rights reserved 2092-6413/16 www.nature.com/emm ORIGINAL ARTICLE Integrated computational approach to the analysis of RNA-seq data reveals new transcriptional regulators of psoriasis Alena Zolotarenko1, Evgeny Chekalin1, Alexandre Mesentsev1, Ludmila Kiseleva2, Elena Gribanova2, Rohini Mehta3, Ancha Baranova3,4,5,6, Tatiana V Tatarinova6,7,8, Eleonora S Piruzian1 and Sergey Bruskin1,5 Psoriasis is a common inflammatory skin disease with complex etiology and chronic progression. To provide novel insights into the regulatory molecular mechanisms of the disease, we performed RNA sequencing analysis of 14 pairs of skin samples collected from patients with psoriasis. Subsequent pathway analysis and extraction of the transcriptional regulators governing psoriasis-associated pathways was executed using a combination of the MetaCore Interactome enrichment tool and the cisExpress algorithm, followed by comparison to a set of previously described psoriasis response elements. A comparative approach allowed us to identify 42 core transcriptional regulators of the disease associated with inflammation (NFκB, IRF9, JUN, FOS, SRF), the activity of T cells in psoriatic lesions (STAT6, FOXP3, NFATC2, GATA3, TCF7, RUNX1), the hyper- proliferation and migration of keratinocytes (JUN, FOS, NFIB, TFAP2A, TFAP2C) and lipid metabolism (TFAP2, RARA, VDR). In addition to the core regulators, we identified 38 transcription factors previously not associated with the disease that can clarify the pathogenesis of psoriasis. To illustrate these findings, we analyzed the regulatory role of one of the identified transcription factors (TFs), FOXA1. Using ChIP-seq and RNA-seq data, we concluded that the atypical expression of the FOXA1 TF is an important player in the disease as it inhibits the maturation of naive T cells into the (CD4+FOXA1+CD47+CD69+PD-L1(hi) FOXP3 − ) regulatory T cell subpopulation, therefore contributing to the development of psoriatic skin lesions. -

Functional Analysis of Notch Signaling During Vertebrate Retinal Development
Functional Analysis of Notch Signaling during Vertebrate Retinal Development The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Mizeracka, Karolina. 2012. Functional Analysis of Notch Signaling during Vertebrate Retinal Development. Doctoral dissertation, Harvard University. Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:10344746 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA © 2012 - Karolina Mizeracka All rights reserved. Advisor: Constance L. Cepko Karolina Mizeracka Functional analysis of Notch signaling during vertebrate retinal development Abstract The process of cell fate determination, which establishes the vastly diverse set of neural cell types found in the central nervous system, remains poorly understood. During retinal development, multipotent retinal progenitor cells generate seven major cell types, including photoreceptors, interneurons, and glia, in an ordered temporal sequence. The behavior of these progenitor cells is influenced by the Notch pathway, a widely utilized signal during embryogenesis which can regulate proliferation and cell fate decisions. To examine the underlying genetic changes that occur when Notch1 is removed from individual retinal cells, microarray analysis of single cells from wild type or Notch1 conditional knockout retinas was performed. Notch1 deficient cells downregulated progenitor and cell cycle marker genes, while robustly upregulating genes associated with rod genesis. Single wild type cells expressed markers of both rod photoreceptors and interneurons, suggesting that these cells were in a transitional state. -

Specification of the Hair Follicle Niche Occurs Prior to Its Formation and Is Progenitor Dependent
bioRxiv preprint doi: https://doi.org/10.1101/414839; this version posted September 12, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Mok, Saxena, Heitman et al., 2018 Fate Before Function: Specification of the Hair Follicle Niche Occurs Prior to its Formation and Is Progenitor Dependent Ka-Wai Mok,1,2,9 Nivedita Saxena,1,2,3,9 Nicholas Heitman,1,2,3,9 Laura Grisanti,1,2 Devika Srivastava,1,2 Mauro Muraro,4 Tina Jacob,5 Rachel Sennett,1,2 Zichen Wang,6 Yutao Su,7 Lu M. Yang,7 Avi Ma’ayan,6 David M. Ornitz7, Maria Kasper,5 and Michael Rendl1,2,3,8,10,* 1Black Family Stem Cell Institute, Icahn School of Medicine at Mount Sinai, Atran Building AB7-10C, Box 1020; 1428 Madison Ave, New York, NY 10029, USA 2Department of Cell, Developmental and Regenerative Biology, Icahn School of Medicine at Mount Sinai, Atran Building AB7-10C, Box 1020; 1428 Madison Ave, New York, NY 10029, USA 3Graduate School of Biomedical Sciences; Icahn School of Medicine at Mount Sinai, Atran Building AB7-10C, Box 1020; 1428 Madison Ave, New York, NY 10029, USA 4Oncode Institute, Hubrecht Institute–KNAW (Royal Netherlands Academy of Arts and Sciences) and University Medical Center Utrecht, 3584 CT Utrecht, the Netherlands 5Department of Biosciences and Nutrition and Center for Innovative Medicine, Karolinska Institutet. 141 83 Huddinge, Sweden 6Department of Pharmacological Sciences, Mount Sinai Center for Bioinformatics, BD2K-LINCS Data Coordination and Integration Center, Knowledge Management Center for Illuminating the Druggable Genome (KMC-IDG), Icahn School of Medicine at Mount Sinai, One Gustave L. -

Development Multiple Checkpoints in Invariant NKT Cell Essential
Essential Functions for ID Proteins at Multiple Checkpoints in Invariant NKT Cell Development This information is current as Mihalis Verykokakis, Veena Krishnamoorthy, Antonio of September 27, 2021. Iavarone, Anna Lasorella, Mikael Sigvardsson and Barbara L. Kee J Immunol 2013; 191:5973-5983; Prepublished online 15 November 2013; doi: 10.4049/jimmunol.1301521 Downloaded from http://www.jimmunol.org/content/191/12/5973 Supplementary http://www.jimmunol.org/content/suppl/2013/11/18/jimmunol.130152 Material 1.DC1 http://www.jimmunol.org/ References This article cites 52 articles, 22 of which you can access for free at: http://www.jimmunol.org/content/191/12/5973.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision by guest on September 27, 2021 • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Essential Functions for ID Proteins at Multiple Checkpoints in Invariant NKT Cell Development Mihalis Verykokakis,*,† Veena Krishnamoorthy,* Antonio Iavarone,‡,x,{ Anna Lasorella,‡,x,{ Mikael Sigvardsson,‖ and Barbara L. -

Response of Adult Mouse Uterus to Early Disruption of Estrogen Receptor-A Signaling Is Influenced by Kru¨Ppel-Like Factor 9
147 Response of adult mouse uterus to early disruption of estrogen receptor-a signaling is influenced by Kru¨ppel-like factor 9 C D Simmons, J M P Pabona, Z Zeng, M C Velarde, D Gaddy, F A Simmen and R C M Simmen1 Department of Physiology and Biophysics, University of Arkansas for Medical Sciences, 4301 W Markham Street, Little Rock, Arkansas 72202, USA 1Arkansas Children’s Nutrition Center, 15 Children’s Way, Little Rock, Arkansas 72202, USA (Correspondence should be addressed to R C M Simmen; Email: [email protected]) (M C Velarde is now at the Buck Institute for Age Research, Novato, California 94945, USA) Abstract Inappropriate early exposure of the hormone-responsive (PNDs) 1–5. Uterine tissues collected at PND84 were uterus to estrogenic compounds is associated with increased subjected to histological, immunological, and molecular risk for adult reproductive diseases including endometrial analyses. Compared with WT mice, KO mice demonstrated cancers. While the dysregulation of estrogen receptor-a larger endometrial glands and lower endometrial gland (ESR1) signaling is well acknowledged to mediate early numbers; DES exposure exacerbated these differences. Loss events in tumor initiation, mechanisms contributing to of KLF9 expression resulted in increased glandular ESR1 sustained ESR1 activity later in life and leading to induction immunoreactivity with DES, without effects on serum of oncogenic pathways remain poorly understood. We had estradiol levels. Quantitative RT-PCR analyses indicated shown previously that the transcription factor Kru¨ppel-like altered expression of uterine genes commonly dysregulated in factor 9 (KLF9) represses ESR1 expression and activity in endometrial cancers (Akt1, Mmp9, Slpi, and Tgfb1) and of Ishikawa endometrial glandular epithelial cells. -

Epstein–Barr Virus Nuclear Antigen 3C Regulated Genes in Lymphoblastoid Cell Lines
Epstein–Barr virus nuclear antigen 3C regulated genes in lymphoblastoid cell lines Bo Zhaoa,1, Jessica C. Marb,c,1, Seiji Maruod,1, Sungwook Leea, Benjamin E. Gewurza, Eric Johannsena, Kristina Holtonb,c, Renee Rubiob,c, Kenzo Takadad, John Quackenbushb,c,2, and Elliott Kieffa,2 aDepartment of Medicine and Microbiology and Molecular Genetics, Channing Laboratory, Brigham and Women’s Hospital and Harvard Medical School, Boston, MA 02115; bDepartment of Biostatistics, Harvard School of Public Health, Boston, MA 02115; cDepartment of Biostatistics and Computational Biology and Department of Cancer Biology, Dana-Farber Cancer Institute, Boston, MA 02115; and dDepartment of Tumor Virology, Institute for Genetic Medicine, Hokkaido University, Sapporo 060-0815, Japan Contributed by Elliott Kieff, November 23, 2010 (sent for review September 29, 2010) EBV nuclear antigen 3C (EBNA3C) is an essential transcription factor sequence-specific transcription factor that also mediates EBNA2 for EBV transformed lymphoblast cell line (LCL) growth. To identify and Notch association with DNA. Therefore, EBNA3C associa- EBNA3C-regulated genes in LCLs, microarrays were used to measure tion with RBPJ in LCLs may affect EBNA2-, EBNA3A-, RNA abundances in each of three different LCLs that conditionally EBNA3B-, or Notch-mediated transcription (5–8). For example, express EBNA3C fused to a 4-OH-Tamoxifen–dependent estrogen experimental two- to threefold EBNA3A overexpression decrea- receptor hormone binding domain (EBNA3CHT). At least three RNAs ses EBNA2 interaction with RBPJ, interrupts EBNA2 up-regu- were assayed for each EBNA3CHT LCL under nonpermissive condi- lation of MYC, and stops LCL growth (9). Also, activated Notch tions, permissive conditions, and nonpermissive conditions with expression can partially substitute for EBNA2 in maintaining LCL wild-type EBNA3C transcomplementation. -

T Cells + IL-4 Expression in CD4 KLF13 Cooperates with C-Maf to Regulate
KLF13 Cooperates with c-Maf To Regulate IL-4 Expression in CD4 + T Cells Seok Joo Kwon, Juan Crespo-Barreto, Wei Zhang, Tianhong Wang, Dong Seok Kim, Alan Krensky and Carol Clayberger This information is current as of September 26, 2021. J Immunol published online 12 May 2014 http://www.jimmunol.org/content/early/2014/05/10/jimmun ol.1302830 Downloaded from Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision http://www.jimmunol.org/ • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription by guest on September 26, 2021 Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Published May 12, 2014, doi:10.4049/jimmunol.1302830 The Journal of Immunology KLF13 Cooperates with c-Maf To Regulate IL-4 Expression in CD4+ T Cells Seok Joo Kwon,*,1 Juan Crespo-Barreto,*,1 Wei Zhang,*,1 Tianhong Wang,* Dong Seok Kim,* Alan Krensky,*,†,‡ and Carol Clayberger*,† Kruppel-like factor (KLF) 13 is a transcription factor that positively regulates expression of the chemokine RANTES 3–5 d after activation of T cells. -

Gene Expression Profiling of Glucocorticoid-Inhibited Osteoblasts
175 Gene expression profiling of glucocorticoid-inhibited osteoblasts Nathalie Leclerc1, Cynthia A Luppen1, Vincent V Ho1, Shailender Nagpal1, Joseph G Hacia1, Elisheva Smith2 and Baruch Frenkel1,2 1Department of Biochemistry and Molecular Biology, Institute for Genetic Medicine, Keck School of Medicine at the University of Southern California, 2250 Alcazar Street, Los Angeles, California 90033, USA 2Department of Orthopedic Surgery, Institute for Genetic Medicine, Keck School of Medicine at the University of Southern California, 2250 Alcazar Street, Los Angeles, California 90033, USA (Requests for offprints should be addressed to Baruch Frenkel; Email: [email protected]) Abstract Glucocorticoid (GC) treatment for the management of autoimmune and inflammatory diseases is associated with decreased bone formation and increased risk for fracture. In MC3T3-E1 cell cultures, 0·1 – 1 µM dexamethasone (DEX) arrests development of the osteoblast phenotype when administration commences at a commitment stage around the time of confluency. To gain new insights into GC-induced osteoporosis, we performed microarray-based gene expression analysis of GC-arrested MC3T3-E1 cultures, 2·5 days after the administration of DEX. Of the .12 000 transcripts interrogated, 74 were up-regulated and 17 were down-regulated by at least 2·5-fold (P¯0·05). Some of these genes, such as Mmp13, Serum/GC-regulated kinase and Tieg, have previously been reported as GC-responsive. Others are shown here for the first time to respond to GCs. DEX strongly repressed Krox20/Egr2 at both