Identification of KLF13 Interacting Partners in the Heart
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Kruppel-Like Factor 9 Inhibits Glioblastoma Stemness
KRUPPEL-LIKE FACTOR 9 INHIBITS GLIOBLASTOMA STEMNESS THROUGH GLOBAL TRANSCRIPTION REPRESSION AND INHIBITION OF INTEGRIN ALPHA 6 AND CD151 By Jessica Tilghman A dissertation submitted to Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy Baltimore, Maryland October, 2015 Abstract Glioblastoma (GBM) stem cells (GSCs) represent tumor-propagating cells with stem-like characteristics (stemness) that contribute disproportionately to GBM drug resistance and tumor recurrence. Understanding the mechanisms supporting GSC stemness is important for developing novel strategies that target tumor propagation to inhibit cancer progression and improve patient survival. Krüppel-like factor 9 (KLF9) has emerged as a regulator of cell differentiation, neural development, and oncogenesis; however, the molecular basis for KLF9’s diverse contextual functions has been unclear. We establish for the first time a genome-wide map of KLF9-regulated targets in human glioblastoma stem-like cells, and show that KLF9 functions as a transcriptional repressor and thereby regulates multiple signaling pathways involved in oncogenesis and regulation of cancer stem-like phenotype. A detailed analysis of two novel KLF9 targets suggests that KLF9 inhibits glioma cell stemness by repressing expression of integrin α6 and CD151. The expression of one candidate KLF9 target gene ITGA6 coding for integrin α6 was verified to be downregulated by KLF9 in GSCs. ITGA6 transcription repression by KLF9 altered GBM neurosphere cell behavior as evidenced by reduced cell adhesion to and migration through membrane coated with the integrin α6 ligand laminin. Forced expression of integrin α6 partially rescued GBM neurosphere cells from the differentiating and adhesion/migration-inhibiting effects of KLF9. -

Reversal of Glucocorticoid Resistance in Paediatric Acute Lymphoblastic Leukaemia Is Dependent on Restoring BIM Expression
www.nature.com/bjc ARTICLE Translational Therapeutics Reversal of glucocorticoid resistance in paediatric acute lymphoblastic leukaemia is dependent on restoring BIM expression Cara E. Toscan1, Duohui Jing1, Chelsea Mayoh1 and Richard B. Lock1 BACKGROUND: Acute lymphoblastic leukaemia (ALL) is the most common paediatric malignancy. Glucocorticoids form a critical component of chemotherapy regimens and resistance to glucocorticoid therapy is predictive of poor outcome. We have previously shown that glucocorticoid resistance is associated with upregulation of the oncogene C-MYC and failure to induce the proapoptotic gene BIM. METHODS: A high-throughput screening (HTS) campaign was carried out to identify glucocorticoid sensitisers against an ALL xenograft derived from a glucocorticoid-resistant paediatric patient. Gene expression analysis was carried out using Illumina microarrays. Efficacy, messenger RNA and protein analysis were carried out by Resazurin assay, reverse transcription-PCR and immunoblotting, respectively. RESULTS: A novel glucocorticoid sensitiser, 2-((4,5-dihydro-1H-imidazol-2-yl)thio)-N-isopropyl-N-phenylacetamide (GCS-3), was identified from the HTS campaign. The sensitising effect was specific to glucocorticoids and synergy was observed in a range of dexamethasone-resistant and dexamethasone-sensitive xenografts representative of B-ALL, T-ALL and Philadelphia chromosome- positive ALL. GCS-3 in combination with dexamethasone downregulated C-MYC and significantly upregulated BIM expression in a glucocorticoid-resistant ALL xenograft. The GCS-3/dexamethasone combination significantly increased binding of the glucocorticoid receptor to a novel BIM enhancer, which is associated with glucocorticoid sensitivity. CONCLUSIONS: This study describes the potential of the novel glucocorticoid sensitiser, GCS-3, as a biological tool to interrogate glucocorticoid action and resistance. -

Tgfβ-Regulated Gene Expression by Smads and Sp1/KLF-Like Transcription Factors in Cancer VOLKER ELLENRIEDER
ANTICANCER RESEARCH 28 : 1531-1540 (2008) Review TGFβ-regulated Gene Expression by Smads and Sp1/KLF-like Transcription Factors in Cancer VOLKER ELLENRIEDER Signal Transduction Laboratory, Internal Medicine, Department of Gastroenterology and Endocrinology, University of Marburg, Marburg, Germany Abstract. Transforming growth factor beta (TGF β) controls complex induces the canonical Smad signaling molecules which vital cellular functions through its ability to regulate gene then translocate into the nucleus to regulate transcription (2). The expression. TGFβ binding to its transmembrane receptor cellular response to TGF β can be extremely variable depending kinases initiates distinct intracellular signalling cascades on the cell type and the activation status of a cell at a given time. including the Smad signalling and transcription factors and also For instance, TGF β induces growth arrest and apoptosis in Smad-independent pathways. In normal epithelial cells, TGF β healthy epithelial cells, whereas it can also promote tumor stimulation induces a cytostatic program which includes the progression through stimulation of cell proliferation and the transcriptional repression of the c-Myc oncogene and the later induction of an epithelial-to-mesenchymal transition of tumor induction of the cell cycle inhibitors p15 INK4b and p21 Cip1 . cells (1, 3). In the last decade it has become clear that both the During carcinogenesis, however, many tumor cells lose their tumor suppressing and the tumor promoting functions of TGF β ability to respond to TGF β with growth inhibition, and instead, are primarily regulated on the level of gene expression through activate genes involved in cell proliferation, invasion and Smad-dependent and -independent mechanisms (1, 2, 4). -

Using Viper, a Package for Virtual Inference of Protein-Activity by Enriched Regulon Analysis
Using viper, a package for Virtual Inference of Protein-activity by Enriched Regulon analysis Mariano J. Alvarez1,2, Federico M. Giorgi1, and Andrea Califano1 1Department of Systems Biology, Columbia University, 1130 St. Nicholas Ave., New York 2DarwinHealth Inc, 3960 Broadway, New York May 19, 2021 1 Overview of VIPER Phenotypic changes effected by pathophysiological events are now routinely captured by gene expression pro- file (GEP) measurements, determining mRNA abundance on a genome-wide scale in a cellular population[8, 9]. In contrast, methods to measure protein abundance on a proteome-wide scale using arrays[11] or mass spectrometry[10] technologies are far less developed, covering only a fraction of proteins, requiring large amounts of tissue, and failing to directly capture protein activity. Furthermore, mRNA expression does not constitute a reliable predictor of protein activity, as it fails to capture a variety of post-transcriptional and post-translational events that are involved in its modulation. Even reliable measurements of protein abundance, for instance by low-throughput antibody based methods or by higher-throughput methods such as mass spectrometry, do not necessarily provide quantitative assessment of functional activity. For instance, enzymatic activity of signal transduction proteins, such as kinases, ubiquitin ligases, and acetyltransferases, is frequently modulated by post-translational modification events that do not affect total protein abundance. Similarly, transcription factors may require post-translationally mediated activation, nuclear translocation, and co-factor availability before they may regulate specific repertoires of their transcriptional targets. Fi- nally, most target-specific drugs affect the activity of their protein substrates rather than their protein or mRNA transcript abundance. -

GATA2 Regulates Mast Cell Identity and Responsiveness to Antigenic Stimulation by Promoting Chromatin Remodeling at Super- Enhancers
ARTICLE https://doi.org/10.1038/s41467-020-20766-0 OPEN GATA2 regulates mast cell identity and responsiveness to antigenic stimulation by promoting chromatin remodeling at super- enhancers Yapeng Li1, Junfeng Gao 1, Mohammad Kamran1, Laura Harmacek2, Thomas Danhorn 2, Sonia M. Leach1,2, ✉ Brian P. O’Connor2, James R. Hagman 1,3 & Hua Huang 1,3 1234567890():,; Mast cells are critical effectors of allergic inflammation and protection against parasitic infections. We previously demonstrated that transcription factors GATA2 and MITF are the mast cell lineage-determining factors. However, it is unclear whether these lineage- determining factors regulate chromatin accessibility at mast cell enhancer regions. In this study, we demonstrate that GATA2 promotes chromatin accessibility at the super-enhancers of mast cell identity genes and primes both typical and super-enhancers at genes that respond to antigenic stimulation. We find that the number and densities of GATA2- but not MITF-bound sites at the super-enhancers are several folds higher than that at the typical enhancers. Our studies reveal that GATA2 promotes robust gene transcription to maintain mast cell identity and respond to antigenic stimulation by binding to super-enhancer regions with dense GATA2 binding sites available at key mast cell genes. 1 Department of Immunology and Genomic Medicine, National Jewish Health, Denver, CO 80206, USA. 2 Center for Genes, Environment and Health, National Jewish Health, Denver, CO 80206, USA. 3 Department of Immunology and Microbiology, University of Colorado Anschutz Medical Campus, Aurora, ✉ CO 80045, USA. email: [email protected] NATURE COMMUNICATIONS | (2021) 12:494 | https://doi.org/10.1038/s41467-020-20766-0 | www.nature.com/naturecommunications 1 ARTICLE NATURE COMMUNICATIONS | https://doi.org/10.1038/s41467-020-20766-0 ast cells (MCs) are critical effectors in immunity that at key MC genes. -

Forced Activation of Notch in Macrophages Represses Tumor Growth By
Author Manuscript Published OnlineFirst on January 12, 2016; DOI: 10.1158/0008-5472.CAN-15-2019 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Forced Activation of Notch in Macrophages Represses Tumor Growth by Upregulating miR-125a and Disabling Tumor-Associated Macrophages Jun-Long Zhao1, Fei Huang1, Fei He2, Chun-Chen Gao1, Shi-Qian Liang1, Peng-Fei Ma2, Guang-Ying Dong1, Hua Han1, 2, Hong-Yan Qin1 1State Key Laboratory of Cancer Biology, Department of Medical Genetics and Developmental Biology, 2Department of Hepatic Surgery, Xijing Hospital, Fourth Military Medical University, Xi’an 710032, China Corresponding Authors: Hong-Yan Qin: Department of Medical Genetics and Developmental Biology, Fourth Military Medical University, Chang-Le Xi Street #169, Xi’an 710032, China; Tel.: +86 29 84774487; Fax: +86 29 83246270; Email: [email protected] Hua Han: Department of Medical Genetics and Developmental Biology, Fourth Military Medical University, Chang-Le Xi Street #169, Xi’an 710032, China; Tel.: +86 29 84774513, Fax: +86 29 83246270; Email: [email protected] Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/). J-L. Zhao and F. Huang contributed equally to this study. Running title: Notch regulates TAMs through miR-125a Disclosure of Potential Conflicts of Interest: The authors disclose no potential conflicts of interest. 1 Downloaded from cancerres.aacrjournals.org on September 28, 2021. © 2016 American Association for Cancer Research. Author Manuscript Published OnlineFirst on January 12, 2016; DOI: 10.1158/0008-5472.CAN-15-2019 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. -
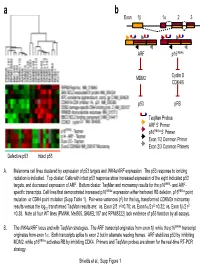
Taqman Probes ARF 5' Primer P16ink4a 5' Primer Exon 1/2
a b Exon 1β 1α 23 ARF p16INK4a MDM2 Cyclin D CDK4/6 p53 pRB TaqMan Probes ARF 5’ Primer p16INK4a 5’ Primer Exon 1/2 Common Primer Exon 2/3 Common Primers Defective p53 Intact p53 A. Melanoma cell lines clustered by expression of p53 targets and INK4a/ARF expression. The p53 response to ionizing radiation is indicated. Top cluster: Cells with intact p53 response show increased expression of the eight indicated p53 targets, and decreased expression of ARF. Bottom cluster: TaqMan and microarray results for the p16INK4- and ARF- specific transcripts. Cell lines that demonstrated increased p16INK4a expression either harbored RB deletion, p16INK4a point 2 mutation or CDK4 point mutation (Supp Table 1). Pair-wise variances (r ) for the log2 transformed CDKN2a microarray 2 2 2 results versus the log10 transformed TaqMan results are: vs. Exon 2/3 r =0.78; vs. Exon1α/2 r =0.32; vs. Exon 1β/2 r =0.38. Note all four WT lines (PMWK, Mel505, SKMEL187 and RPMI8322) lack evidence of p53 function by all assays. B. The INK4a/ARF locus and with TaqMan strategies. The ARF transcript originates from exon 1β while the p16INK4a transcript originates from exon 1α. Both transcripts splice to exon 2 but in alternate reading frames. ARF stabilizes p53 by inhibiting MDM2, while p16INK4a activates RB by inhibiting CDK4. Primers and TaqMan probes are shown for the real-time RT-PCR strategy. Shields et al., Supp Figure 1 SKMEL 28 U01 24h SKMEL WM2664 U01 48h WM2664 U01 24h 24 U01 48h SKMEL 24 U01 24h SKMEL 24 Untreated SKMEL 24 DMSO 48h SKMEL 24 DMSO 24h SKMEL WM2664 DMSO 24h WM2664 DMSO 48h WM2644 Untreated 28 Untreated SKMEL 28 DMSO 48h SKMEL 28 DMSO 24h SKMEL -3.00 -2.00 -1.00 0.00 1.00 2.00 3.00 relative to median expression Genes decreased by UO1 (863) HIF1A Hypoxia-inducible factor 1, alpha subunit NM_001530 RBBP8 Retinoblastoma binding protein 8 NM_002894 Homo sapiens, clone IMAGE:4337652, mRNA BC018676 EIF4EBP1 Eukaryotic translation initiation factor 4E binding protein EXOSC8 Exosome component 8 NM_181503 ENST00000321524 MCM7 MCM7 minichromosome maintenance deficient 7 (S. -

The Expression of Genes Contributing to Pancreatic Adenocarcinoma Progression Is Influenced by the Respective Environment – Sagini Et Al
The expression of genes contributing to pancreatic adenocarcinoma progression is influenced by the respective environment – Sagini et al Supplementary Figure 1: Target genes regulated by TGM2. Figure represents 24 genes regulated by TGM2, which were obtained from Ingenuity Pathway Analysis. As indicated, 9 genes (marked red) are down-regulated by TGM2. On the contrary, 15 genes (marked red) are up-regulated by TGM2. Supplementary Table 1: Functional annotations of genes from Suit2-007 cells growing in pancreatic environment Categoriesa Diseases or p-Valuec Predicted Activation Number of genesf Functions activationd Z-scoree Annotationb Cell movement Cell movement 1,56E-11 increased 2,199 LAMB3, CEACAM6, CCL20, AGR2, MUC1, CXCL1, LAMA3, LCN2, COL17A1, CXCL8, AIF1, MMP7, CEMIP, JUP, SOD2, S100A4, PDGFA, NDRG1, SGK1, IGFBP3, DDR1, IL1A, CDKN1A, NREP, SEMA3E SERPINA3, SDC4, ALPP, CX3CL1, NFKBIA, ANXA3, CDH1, CDCP1, CRYAB, TUBB2B, FOXQ1, SLPI, F3, GRINA, ITGA2, ARPIN/C15orf38- AP3S2, SPTLC1, IL10, TSC22D3, LAMC2, TCAF1, CDH3, MX1, LEP, ZC3H12A, PMP22, IL32, FAM83H, EFNA1, PATJ, CEBPB, SERPINA5, PTK6, EPHB6, JUND, TNFSF14, ERBB3, TNFRSF25, FCAR, CXCL16, HLA-A, CEACAM1, FAT1, AHR, CSF2RA, CLDN7, MAPK13, FERMT1, TCAF2, MST1R, CD99, PTP4A2, PHLDA1, DEFB1, RHOB, TNFSF15, CD44, CSF2, SERPINB5, TGM2, SRC, ITGA6, TNC, HNRNPA2B1, RHOD, SKI, KISS1, TACSTD2, GNAI2, CXCL2, NFKB2, TAGLN2, TNF, CD74, PTPRK, STAT3, ARHGAP21, VEGFA, MYH9, SAA1, F11R, PDCD4, IQGAP1, DCN, MAPK8IP3, STC1, ADAM15, LTBP2, HOOK1, CST3, EPHA1, TIMP2, LPAR2, CORO1A, CLDN3, MYO1C, -

Circptpra Acts As a Tumor Suppressor in Bladder Cancer by Sponging Mir-636 and Upregulating KLF9
www.aging-us.com AGING 2019, Vol. 11, No. 23 Research Paper CircPTPRA acts as a tumor suppressor in bladder cancer by sponging miR-636 and upregulating KLF9 Qingqing He1,2,*, Lifang Huang2,*, Dong Yan1,2,*, Junming Bi1,2, Meihua Yang1,2, Jian Huang1,2, Tianxin Lin1,2 1Department of Urology, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, China 2Guangdong Provincial Key Laboratory of Malignant Tumor Epigenetics and Gene Regulation, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, China *Equal contribution Correspondence to: Jian Huang, Tianxin Lin; email: [email protected], [email protected] Keywords: bladder cancer, circPTPRA, miR-636, KLF9, proliferation Received: September 4, 2019 Accepted: November 18, 2019 Published: December 10, 2019 Copyright: He et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Growing evidence suggests that circular RNAs (circRNAs) play pivotal roles in cancer progression. In this study, bioinformatic analysis identified a dysregulated circRNA termed circPTPRA in bladder cancer (BC). By using qRT- PCR analysis, we verified that circPTPRA is down-regulated in clinical BC specimens compared with the matched non-tumor samples, while correlation analyses showed that low circPTPRA expression is associated with poor prognosis, advanced tumor stage and larger tumor size. Based on these findings, we conducted functional assays and revealed that circPTPRA inhibits BC cell proliferation in vitro and tumor growth in vivo. In addition, RNA pull-down, miRNA capture, FISH, and luciferase reporter assays demonstrated that circPTPRA can directly sponge miR-636. -

Microrna-200B Regulates the Proliferation and Differentiation of Ovine Preadipocytes by Targeting P27 and KLF9
animals Article MicroRNA-200b Regulates the Proliferation and Differentiation of Ovine Preadipocytes by Targeting p27 and KLF9 Xiayang Jin , Jiqing Wang * , Jiang Hu, Xiu Liu, Shaobin Li , Yujie Lu, Huimin Zhen, Mingna Li, Zhidong Zhao and Yuzhu Luo * Gansu Key Laboratory of Herbivorous Animal Biotechnology, Faculty of Animal Science and Technology, Gansu Agricultural University, Lanzhou 730070, China; [email protected] (X.J.); [email protected] (J.H.); [email protected] (X.L.); [email protected] (S.L.); [email protected] (Y.L.); [email protected] (H.Z.); [email protected] (M.L.); [email protected] (Z.Z.) * Correspondence: [email protected] (J.W.); [email protected] (Y.L.); Tel.: +86-931-763-2469 (J.W.); +86-931-763-2483 (Y.L.) Simple Summary: The miR-200b has been shown to play an important role in preadipocyte prolifer- ation and differentiation. Herein, we explored the role of miR-200b in ovine adipocyte development, using Oil Red O staining, cell viability analysis, EdU and RT-qPCR. The results showed that miR-200b facilitated proliferation and suppressed the differentiation of preadipocytes. The dual fluorescent reporter vector experiments showed that miR-200b directly targeted p27 and KLF9. Meanwhile, we demonstrated that p27 significantly inhibited the proliferation, while KLF9 significantly promoted the differentiation of preadipocytes. Abstract: MicroRNAs (miRNAs) are crucial regulatory molecules in lipid deposition and metabolism. Citation: Jin, X.; Wang, J.; Hu, J.; Liu, However, the effect of miR-200b on the regulation of proliferation and adipogenesis of ovine X.; Li, S.; Lu, Y.; Zhen, H.; Li, M.; Zhao, Z.; Luo, Y. -

Integrated Computational Approach to the Analysis of RNA-Seq Data Reveals New Transcriptional Regulators of Psoriasis
OPEN Experimental & Molecular Medicine (2016) 48, e268; doi:10.1038/emm.2016.97 & 2016 KSBMB. All rights reserved 2092-6413/16 www.nature.com/emm ORIGINAL ARTICLE Integrated computational approach to the analysis of RNA-seq data reveals new transcriptional regulators of psoriasis Alena Zolotarenko1, Evgeny Chekalin1, Alexandre Mesentsev1, Ludmila Kiseleva2, Elena Gribanova2, Rohini Mehta3, Ancha Baranova3,4,5,6, Tatiana V Tatarinova6,7,8, Eleonora S Piruzian1 and Sergey Bruskin1,5 Psoriasis is a common inflammatory skin disease with complex etiology and chronic progression. To provide novel insights into the regulatory molecular mechanisms of the disease, we performed RNA sequencing analysis of 14 pairs of skin samples collected from patients with psoriasis. Subsequent pathway analysis and extraction of the transcriptional regulators governing psoriasis-associated pathways was executed using a combination of the MetaCore Interactome enrichment tool and the cisExpress algorithm, followed by comparison to a set of previously described psoriasis response elements. A comparative approach allowed us to identify 42 core transcriptional regulators of the disease associated with inflammation (NFκB, IRF9, JUN, FOS, SRF), the activity of T cells in psoriatic lesions (STAT6, FOXP3, NFATC2, GATA3, TCF7, RUNX1), the hyper- proliferation and migration of keratinocytes (JUN, FOS, NFIB, TFAP2A, TFAP2C) and lipid metabolism (TFAP2, RARA, VDR). In addition to the core regulators, we identified 38 transcription factors previously not associated with the disease that can clarify the pathogenesis of psoriasis. To illustrate these findings, we analyzed the regulatory role of one of the identified transcription factors (TFs), FOXA1. Using ChIP-seq and RNA-seq data, we concluded that the atypical expression of the FOXA1 TF is an important player in the disease as it inhibits the maturation of naive T cells into the (CD4+FOXA1+CD47+CD69+PD-L1(hi) FOXP3 − ) regulatory T cell subpopulation, therefore contributing to the development of psoriatic skin lesions. -

Functional Analysis of Notch Signaling During Vertebrate Retinal Development
Functional Analysis of Notch Signaling during Vertebrate Retinal Development The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Mizeracka, Karolina. 2012. Functional Analysis of Notch Signaling during Vertebrate Retinal Development. Doctoral dissertation, Harvard University. Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:10344746 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA © 2012 - Karolina Mizeracka All rights reserved. Advisor: Constance L. Cepko Karolina Mizeracka Functional analysis of Notch signaling during vertebrate retinal development Abstract The process of cell fate determination, which establishes the vastly diverse set of neural cell types found in the central nervous system, remains poorly understood. During retinal development, multipotent retinal progenitor cells generate seven major cell types, including photoreceptors, interneurons, and glia, in an ordered temporal sequence. The behavior of these progenitor cells is influenced by the Notch pathway, a widely utilized signal during embryogenesis which can regulate proliferation and cell fate decisions. To examine the underlying genetic changes that occur when Notch1 is removed from individual retinal cells, microarray analysis of single cells from wild type or Notch1 conditional knockout retinas was performed. Notch1 deficient cells downregulated progenitor and cell cycle marker genes, while robustly upregulating genes associated with rod genesis. Single wild type cells expressed markers of both rod photoreceptors and interneurons, suggesting that these cells were in a transitional state.