The Discovery, Distribution and Diversity of DNA Viruses Associated with Drosophila Melanogaster in Europe Authors: Megan A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Using Cell Lines to Study Factors Affecting Transmission of Fish Viruses
Using cell lines to study factors affecting transmission of fish viruses by Phuc Hoang Pham A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree of Doctor of Philosophy in Biology Waterloo, Ontario, Canada, 2014 ©Phuc Hoang Pham 2014 AUTHOR'S DECLARATION I hereby declare that I am the sole author of this thesis. This is a true copy of the thesis, including any required final revisions, as accepted by my examiners. I understand that my thesis may be made electronically available to the public. ii ABSTRACT Factors that can influence the transmission of aquatic viruses in fish production facilities and natural environment are the immune defense of host species, the ability of viruses to infect host cells, and the environmental persistence of viruses. In this thesis, fish cell lines were used to study different aspects of these factors. Five viruses were used in this study: viral hemorrhagic septicemia virus (VHSV) from the Rhabdoviridae family; chum salmon reovirus (CSV) from the Reoviridae family; infectious pancreatic necrosis virus (IPNV) from the Birnaviridae family; and grouper iridovirus (GIV) and frog virus-3 (FV3) from the Iridoviridae family. The first factor affecting the transmission of fish viruses examined in this thesis is the immune defense of host species. In this work, infections of marine VHSV-IVa and freshwater VHSV-IVb were studied in two rainbow trout cell lines, RTgill-W1 from the gill epithelium, and RTS11 from spleen macrophages. RTgill-W1 produced infectious progeny of both VHSV-IVa and -IVb. However, VHSV-IVa was more infectious than IVb toward RTgill-W1: IVa caused cytopathic effects (CPE) at a lower viral titre, elicited CPE earlier, and yielded higher titres. -

Changes to Virus Taxonomy 2004
Arch Virol (2005) 150: 189–198 DOI 10.1007/s00705-004-0429-1 Changes to virus taxonomy 2004 M. A. Mayo (ICTV Secretary) Scottish Crop Research Institute, Invergowrie, Dundee, U.K. Received July 30, 2004; accepted September 25, 2004 Published online November 10, 2004 c Springer-Verlag 2004 This note presents a compilation of recent changes to virus taxonomy decided by voting by the ICTV membership following recommendations from the ICTV Executive Committee. The changes are presented in the Table as decisions promoted by the Subcommittees of the EC and are grouped according to the major hosts of the viruses involved. These new taxa will be presented in more detail in the 8th ICTV Report scheduled to be published near the end of 2004 (Fauquet et al., 2004). Fauquet, C.M., Mayo, M.A., Maniloff, J., Desselberger, U., and Ball, L.A. (eds) (2004). Virus Taxonomy, VIIIth Report of the ICTV. Elsevier/Academic Press, London, pp. 1258. Recent changes to virus taxonomy Viruses of vertebrates Family Arenaviridae • Designate Cupixi virus as a species in the genus Arenavirus • Designate Bear Canyon virus as a species in the genus Arenavirus • Designate Allpahuayo virus as a species in the genus Arenavirus Family Birnaviridae • Assign Blotched snakehead virus as an unassigned species in family Birnaviridae Family Circoviridae • Create a new genus (Anellovirus) with Torque teno virus as type species Family Coronaviridae • Recognize a new species Severe acute respiratory syndrome coronavirus in the genus Coro- navirus, family Coronaviridae, order Nidovirales -

Evidence for Viral Infection in the Copepods Labidocera Aestiva And
University of South Florida Scholar Commons Graduate Theses and Dissertations Graduate School January 2012 Evidence for Viral Infection in the Copepods Labidocera aestiva and Acartia tonsa in Tampa Bay, Florida Darren Stephenson Dunlap University of South Florida, [email protected] Follow this and additional works at: http://scholarcommons.usf.edu/etd Part of the American Studies Commons, Other Oceanography and Atmospheric Sciences and Meteorology Commons, and the Virology Commons Scholar Commons Citation Dunlap, Darren Stephenson, "Evidence for Viral Infection in the Copepods Labidocera aestiva and Acartia tonsa in Tampa Bay, Florida" (2012). Graduate Theses and Dissertations. http://scholarcommons.usf.edu/etd/4032 This Thesis is brought to you for free and open access by the Graduate School at Scholar Commons. It has been accepted for inclusion in Graduate Theses and Dissertations by an authorized administrator of Scholar Commons. For more information, please contact [email protected]. Evidence of Viruses in the Copepods Labidocera aestiva and Acartia tonsa in Tampa Bay, Florida By Darren S. Dunlap A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science College of Marine Science University of South Florida Major Professor: Mya Breitbart, Ph.D Kendra Daly, Ph.D Ian Hewson, Ph.D Date of Approval: March 19, 2012 Key Words: Copepods, Single-stranded DNA Viruses, Mesozooplankton, Transmission Electron Microscopy, Metagenomics Copyright © 2012, Darren Stephenson Dunlap DEDICATION None of this would have been possible without the generous love and support of my entire family over the years. My parents, Steve and Jill Dunlap, have always encouraged my pursuits with support and love, and their persistence of throwing me into lakes and rivers is largely responsible for my passion for Marine Science. -

Characterization of Viral Communities of Biting Midges and Identification
viruses Article Characterization of Viral Communities of Biting Midges and Identification of Novel Thogotovirus Species and Rhabdovirus Genus Sarah Temmam 1, Sonia Monteil-Bouchard 1, Catherine Robert 1, Jean-Pierre Baudoin 1, Masse Sambou 1, Maxence Aubadie-Ladrix 1, Noémie Labas 1, Didier Raoult 1,2, Oleg Mediannikov 1 and Christelle Desnues 1,* 1 Unité de Recherche sur les Maladies Infectieuses et Tropicales Emergentes (URMITE), UM63 CNRS 7278 IRD 198 INSERM U1095, Aix-Marseille Université, Marseille 13005, France; [email protected] (S.T.); [email protected] (S.M.-B.); [email protected] (C.R.); [email protected] (J.-P.B.); [email protected] (M.S.); [email protected] (M.A.-L.); [email protected] (N.L.); [email protected] (D.R.); [email protected] (O.M.). 2 Fondation IHU Méditerranée Infection, Pôle des Maladies Infectieuses et Tropicales Clinique et Biologique, Fédération de Bactériologie-Hygiène-Virologie, Centre Hospitalo-Universitaire Timone, Méditerranée Infection, Assistance Publique–Hôpitaux de Marseille, Marseille 13005, France * Correspondence: [email protected]; Tel.: +33-0-491324630; Fax: +33-0-491387772 Academic Editors: Johnson Mak, Peter Walker and Marcus Thomas Gilbert Received: 21 October 2015; Accepted: 1 March 2016; Published: 11 March 2016 Abstract: More than two thirds of emerging viruses are of zoonotic origin, and among them RNA viruses represent the majority. Ceratopogonidae (genus Culicoides) are well-known vectors of several viruses responsible for epizooties (bluetongue, epizootic haemorrhagic disease, etc.). They are also vectors of the only known virus infecting humans: the Oropouche virus. Female midges usually feed on a variety of hosts, leading to possible transmission of emerging viruses from animals to humans. -

The DNA Virus Invertebrate Iridescent Virus 6 Is a Target of the Drosophila Rnai Machinery
The DNA virus Invertebrate iridescent virus 6 is a target of the Drosophila RNAi machinery Alfred W. Bronkhorsta,1, Koen W. R. van Cleefa,1, Nicolas Vodovarb,2, Ikbal_ Agah Ince_ c,d,e, Hervé Blancb, Just M. Vlakc, Maria-Carla Salehb,3, and Ronald P. van Rija,3 aDepartment of Medical Microbiology, Nijmegen Centre for Molecular Life Sciences, Nijmegen Institute for Infection, Inflammation, and Immunity, Radboud University Nijmegen Medical Centre, 6500 HB Nijmegen, The Netherlands; bViruses and RNA Interference Group, Institut Pasteur, Centre National de la Recherche Scientifique, Unité de Recherche Associée 3015, 75015 Paris, France; cLaboratory of Virology, Wageningen University, 6708 PB Wageningen, The Netherlands; dDepartment of Genetics and Bioengineering, Yeditepe University, Istanbul 34755, Turkey; and eDepartment of Biosystems Engineering, Faculty of Engineering, Giresun University, Giresun 28100, Turkey Edited by Peter Palese, Mount Sinai School of Medicine, New York, NY, and approved October 19, 2012 (received for review April 28, 2012) RNA viruses in insects are targets of an RNA interference (RNAi)- sequently, are hypersensitive to virus infection and succumb more based antiviral immune response, in which viral replication inter- rapidly than their wild-type (WT) controls (11–14). mediates or viral dsRNA genomes are processed by Dicer-2 (Dcr-2) Small RNA cloning and next-generation sequencing provide into viral small interfering RNAs (vsiRNAs). Whether dsDNA virus detailed insights into vsiRNA biogenesis. In several studies in infections are controlled by the RNAi pathway remains to be insects, the polarity of the vsiRNA population deviates strongly determined. Here, we analyzed the role of RNAi in DNA virus from the highly skewed distribution of positive strand (+) over infection using Drosophila melanogaster infected with Invertebrate negative (−) viral RNAs that is generally observed in (+) RNA iridescent virus 6 (IIV-6) as a model. -

And Giant Guitarfish (Rhynchobatus Djiddensis)
VIRAL DISCOVERY IN BLUEGILL SUNFISH (LEPOMIS MACROCHIRUS) AND GIANT GUITARFISH (RHYNCHOBATUS DJIDDENSIS) BY HISTOPATHOLOGY EVALUATION, METAGENOMIC ANALYSIS AND NEXT GENERATION SEQUENCING by JENNIFER ANNE DILL (Under the Direction of Alvin Camus) ABSTRACT The rapid growth of aquaculture production and international trade in live fish has led to the emergence of many new diseases. The introduction of novel disease agents can result in significant economic losses, as well as threats to vulnerable wild fish populations. Losses are often exacerbated by a lack of agent identification, delay in the development of diagnostic tools and poor knowledge of host range and susceptibility. Examples in bluegill sunfish (Lepomis macrochirus) and the giant guitarfish (Rhynchobatus djiddensis) will be discussed here. Bluegill are popular freshwater game fish, native to eastern North America, living in shallow lakes, ponds, and slow moving waterways. Bluegill experiencing epizootics of proliferative lip and skin lesions, characterized by epidermal hyperplasia, papillomas, and rarely squamous cell carcinoma, were investigated in two isolated poopulations. Next generation genomic sequencing revealed partial DNA sequences of an endogenous retrovirus and the entire circular genome of a novel hepadnavirus. Giant Guitarfish, a rajiform elasmobranch listed as ‘vulnerable’ on the IUCN Red List, are found in the tropical Western Indian Ocean. Proliferative skin lesions were observed on the ventrum and caudal fin of a juvenile male quarantined at a public aquarium following international shipment. Histologically, lesions consisted of papillomatous epidermal hyperplasia with myriad large, amphophilic, intranuclear inclusions. Deep sequencing and metagenomic analysis produced the complete genomes of two novel DNA viruses, a typical polyomavirus and a second unclassified virus with a 20 kb genome tentatively named Colossomavirus. -

Wolbachia-Driven Selective Sweep in a Range Expanding Insect Species
Wolbachia-driven selective sweep in a range expanding insect species Junchen Deng Lunds Universitet Giacomo Assandri Istituto Superiore per la Protezione e la Ricerca Ambientale Pallavi Chauhan Lunds Universitet Ryo Futahashi Trukuba Andrea Galimberti University of Milano–Bicocca: Universita degli Studi di Milano-Bicocca Bengt Hansson Lunds Universitet Lesley Lancaster University of Aberdeen Yuma Takahashi Chiba University graduate school of science Erik I Svensson Lund University: Lunds Universitet Anne Duplouy ( anne.duplouy@helsinki. ) University of Helsinki: Helsingin Yliopisto https://orcid.org/0000-0002-7147-5199 Research article Keywords: Endosymbiosis, phylogeography, damsely, mitochondria, genetic diversity Posted Date: February 24th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-150504/v3 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/24 Abstract Background Evolutionary processes can cause strong spatial genetic signatures, such as local loss of genetic diversity, or conicting histories from mitochondrial versus nuclear markers. Investigating these genetic patterns is important, as they may reveal obscured processes and players. The maternally inherited bacterium Wolbachia is among the most widespread symbionts in insects. Wolbachia typically spreads within host species by conferring direct tness benets, or by manipulating its host reproduction to favour infected over uninfected females. Under sucient selective advantage, the mitochondrial haplotype associated with the favoured symbiotic strains will spread (i.e. hitchhike), resulting in low mitochondrial genetic variation across the host species range. The common bluetail damsely (Ischnura elegans: van der Linden, 1820) has recently emerged as a model organism of the genetics and genomic signatures of range expansion during climate change. -

And Γ- Cytoplasmic Actin in Vaccinia Virus Infection
Lights, Camera, Actin: Divergent roles of β- and γ- cytoplasmic actin in vaccinia virus infection NOORUL BISHARA MARZOOK A thesis submitted in fulfillment of requirements for the degree of Doctor of Philosophy FACULTY OF SCIENCE SCHOOL OF MOLECULAR BIOSCIENCE UNIVERSITY OF SYDNEY 2017 i TABLE OF CONTENTS Table of Contents ........................................................................................................... ii Acknowledgements ....................................................................................................... v Declaration ................................................................................................................... vii Abstract ....................................................................................................................... viii List of Figures ................................................................................................................ x List of Publications Arising From This Work.............................................................. xi Abbreviations Used ..................................................................................................... xii Chapter 1: Introduction ............................................................................................... 1 1.1 The Cytoskeleton ............................................................................................................ 2 1.1.1 The Eukaryotic Cytoskeleton ..................................................................................... -
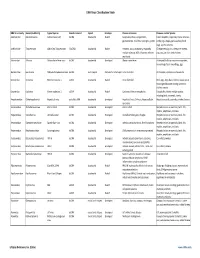
Virus Classification Tables V2.Vd.Xlsx
DNA Virus Classification Table DNA Virus Family Genera (Subfamily) Typical Species Genetic material Capsid Envelope Disease in Humans Diseases in other Species Adenoviridae Mastadenovirus Adenoviruses 1‐47 dsDNA Icosahedral Naked Respiratory illness; conjunctivitis, Canine hepatitis, respiratory illness in horses, gastroenteritis, tonsillitis, meningitis, cystitis cattle, pigs, sheep, goats, sea lions, birds dogs, squirrel enteritis Anelloviridae Torqueviruses Alpha‐Zeta Torqueviruses (‐)ssDNA Icosahedral Naked Hepatitis, lupus, pulmonary, myopathy, Chimpanzee, pig, cow, sheep, tree shrews, multiple sclerosis; 90% of humans infected pigs, cats, sea lions and chickens worldwide Asfarviridae Asfivirus African Swine fever virus dsDNA Icosahedral Enveloped African swine fever Arthropod (tick) transmission or ingestion; hemorrhagic fever in warthogs, pigs Baculoviridae Baculovirus Alpha‐Gamma Baculoviruses dsDNA Stick shaped Occluded or Enveloped none identified Arthropods, Lepidoptera, crustaceans Circoviridae Circovirus Porcine circovirus 1 ssDNA Icosahedral Naked none identified Birds, pigs, dogs; bats; rodents; causes post‐ weaning multisystem wasting syndrome, chicken anemia Circoviridae Cyclovirus Human cyclovirus 1 ssDNA Icosahedral Naked Cyclovirus Vietnam encephalitis Encephalitis; infects multiple species including birds, mammals, insects Hepadnaviridae Orthohepadnavirus Hepatitis B virus partially ssDNA Icosahedral Enveloped Hepatitis B virus; Cirrhosis, Hepatocellular Hepatitis in ducks, squirrels, primates, herons carcinoma Herpesviridae -

Viruses in Transplantation - Not Always Enemies
Viruses in transplantation - not always enemies Virome and transplantation ECCMID 2018 - Madrid Prof. Laurent Kaiser Head Division of Infectious Diseases Laboratory of Virology Geneva Center for Emerging Viral Diseases University Hospital of Geneva ESCMID eLibrary © by author Conflict of interest None ESCMID eLibrary © by author The human virome: definition? Repertoire of viruses found on the surface of/inside any body fluid/tissue • Eukaryotic DNA and RNA viruses • Prokaryotic DNA and RNA viruses (phages) 25 • The “main” viral community (up to 10 bacteriophages in humans) Haynes M. 2011, Metagenomic of the human body • Endogenous viral elements integrated into host chromosomes (8% of the human genome) • NGS is shaping the definition Rascovan N et al. Annu Rev Microbiol 2016;70:125-41 Popgeorgiev N et al. Intervirology 2013;56:395-412 Norman JM et al. Cell 2015;160:447-60 ESCMID eLibraryFoxman EF et al. Nat Rev Microbiol 2011;9:254-64 © by author Viruses routinely known to cause diseases (non exhaustive) Upper resp./oropharyngeal HSV 1 Influenza CNS Mumps virus Rhinovirus JC virus RSV Eye Herpes viruses Parainfluenza HSV Measles Coronavirus Adenovirus LCM virus Cytomegalovirus Flaviviruses Rabies HHV6 Poliovirus Heart Lower respiratory HTLV-1 Coxsackie B virus Rhinoviruses Parainfluenza virus HIV Coronaviruses Respiratory syncytial virus Parainfluenza virus Adenovirus Respiratory syncytial virus Coronaviruses Gastro-intestinal Influenza virus type A and B Human Bocavirus 1 Adenovirus Hepatitis virus type A, B, C, D, E Those that cause -

Finding of Male-Killing Spiroplasma Infecting Drosophila Melanogaster in Africa Implies Transatlantic Migration of This Endosymbiont
Heredity (2006) 97, 27–32 & 2006 Nature Publishing Group All rights reserved 0018-067X/06 $30.00 www.nature.com/hdy Finding of male-killing Spiroplasma infecting Drosophila melanogaster in Africa implies transatlantic migration of this endosymbiont JE Pool, A Wong and CF Aquadro Department of Molecular Biology and Genetics, Cornell University, 233 Biotechnology Building, Ithaca, NY 14853, USA We report the identification of male-killing Spiroplasma in a efficiency appear to increase with female age, and we note wild-caught female Drosophila melanogaster from Uganda, that males born in sex ratio broods display much lower the first such infection to be found in this species outside survivorship than their female siblings. DNA sequence of South America. Among 38 female flies collected from comparisons at three loci suggest that this Spiroplasma Namulonge, Uganda in April, 2005, one produced a total strain is closely related to the male-killing strain previously of 41 female offspring but no males. PCR testing of found to infect D. melanogaster in Brazil, although part of subsequent generations revealed that females retaining one locus appears to show a recombinant history. Implica- Spiroplasma infection continued to produce a large excess tions for the origin and history of male-killing Spiroplasma in of female progeny, while females that had lost Spiroplasma D. melanogaster are discussed. produced offspring with normal sex ratios. Consistent with Heredity (2006) 97, 27–32. doi:10.1038/sj.hdy.6800830; earlier work, we find that male-killing and transmission published online 10 May 2006 Keywords: Spiroplasma; male-killing; Drosophila melanogaster; Africa; migration Introduction few hundred years (David and Capy, 1988). -

Geve: a Genome-Based Endogenous Viral Element Database Provides
Database, 2016, 1–8 doi: 10.1093/database/baw087 Original article Original article Downloaded from https://academic.oup.com/database/article-abstract/doi/10.1093/database/baw087/2630466 by guest on 24 July 2019 gEVE: a genome-based endogenous viral element database provides comprehensive viral protein-coding sequences in mammalian genomes So Nakagawa1,2,* and Mahoko Ueda Takahashi2 1Department of Molecular Life Science, Tokai University School of Medicine, 143 Shimokasuya, Isehara, Kanagawa 259-1193, Japan and 2Micro/Nano Technology Center, Tokai University, 411 Kitakaname, Hiratsuka, Kanagawa, 259-1292, Japan *Corresponding author: Tel: þ81 463 93 1121 ext. 2661; Fax: þ81 463 93 5418; Email: [email protected] Citation details: Nakagawa,S. and Ueda Takahashi,M. gEVE: a genome-based endogenous viral element database pro- vides comprehensive viral protein-coding sequences in mammalian genomes. Database (2016) Vol. 2016: article ID baw087; doi:10.1093/database/baw087 Received 15 December 2015; Revised 15 April 2016; Accepted 4 May 2016 Abstract In mammals, approximately 10% of genome sequences correspond to endogenous viral elements (EVEs), which are derived from ancient viral infections of germ cells. Although most EVEs have been inactivated, some open reading frames (ORFs) of EVEs obtained functions in the hosts. However, EVE ORFs usually remain unannotated in the genomes, and no databases are available for EVE ORFs. To investigate the function and evolution of EVEs in mammalian genomes, we developed EVE ORF databases for 20 genomes of 19 mammalian species. A total of 736,771 non-overlapping EVE ORFs were identified and archived in a database named gEVE (http://geve.med.u-tokai.ac.jp).