Gillessen-Kaesbach–Nishimura Skeletal Dysplasia Due to Pathogenic Variants in ALG9
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Broad and Thematic Remodeling of the Surface Glycoproteome on Isogenic
bioRxiv preprint doi: https://doi.org/10.1101/808139; this version posted October 17, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Broad and thematic remodeling of the surface glycoproteome on isogenic cells transformed with driving proliferative oncogenes Kevin K. Leung1,5, Gary M. Wilson2,5, Lisa L. Kirkemo1, Nicholas M. Riley2,4, Joshua J. Coon2,3, James A. Wells1* 1Department of Pharmaceutical Chemistry, UCSF, San Francisco, CA, USA Departments of Chemistry2 and Biomolecular Chemistry3, University of Wisconsin- Madison, Madison, WI, 53706, USA 4Present address Department of Chemistry, Stanford University, Stanford, CA, 94305, USA 5These authors contributed equally *To whom correspondence should be addressed bioRxiv preprint doi: https://doi.org/10.1101/808139; this version posted October 17, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Abstract: The cell surface proteome, the surfaceome, is the interface for engaging the extracellular space in normal and cancer cells. Here We apply quantitative proteomics of N-linked glycoproteins to reveal how a collection of some 700 surface proteins is dramatically remodeled in an isogenic breast epithelial cell line stably expressing any of six of the most prominent proliferative oncogenes, including the receptor tyrosine kinases, EGFR and HER2, and downstream signaling partners such as KRAS, BRAF, MEK and AKT. -

Yeast Genome Gazetteer P35-65
gazetteer Metabolism 35 tRNA modification mitochondrial transport amino-acid metabolism other tRNA-transcription activities vesicular transport (Golgi network, etc.) nitrogen and sulphur metabolism mRNA synthesis peroxisomal transport nucleotide metabolism mRNA processing (splicing) vacuolar transport phosphate metabolism mRNA processing (5’-end, 3’-end processing extracellular transport carbohydrate metabolism and mRNA degradation) cellular import lipid, fatty-acid and sterol metabolism other mRNA-transcription activities other intracellular-transport activities biosynthesis of vitamins, cofactors and RNA transport prosthetic groups other transcription activities Cellular organization and biogenesis 54 ionic homeostasis organization and biogenesis of cell wall and Protein synthesis 48 plasma membrane Energy 40 ribosomal proteins organization and biogenesis of glycolysis translation (initiation,elongation and cytoskeleton gluconeogenesis termination) organization and biogenesis of endoplasmic pentose-phosphate pathway translational control reticulum and Golgi tricarboxylic-acid pathway tRNA synthetases organization and biogenesis of chromosome respiration other protein-synthesis activities structure fermentation mitochondrial organization and biogenesis metabolism of energy reserves (glycogen Protein destination 49 peroxisomal organization and biogenesis and trehalose) protein folding and stabilization endosomal organization and biogenesis other energy-generation activities protein targeting, sorting and translocation vacuolar and lysosomal -

Congenital Disorders of Glycosylation from a Neurological Perspective
brain sciences Review Congenital Disorders of Glycosylation from a Neurological Perspective Justyna Paprocka 1,* , Aleksandra Jezela-Stanek 2 , Anna Tylki-Szyma´nska 3 and Stephanie Grunewald 4 1 Department of Pediatric Neurology, Faculty of Medical Science in Katowice, Medical University of Silesia, 40-752 Katowice, Poland 2 Department of Genetics and Clinical Immunology, National Institute of Tuberculosis and Lung Diseases, 01-138 Warsaw, Poland; [email protected] 3 Department of Pediatrics, Nutrition and Metabolic Diseases, The Children’s Memorial Health Institute, W 04-730 Warsaw, Poland; [email protected] 4 NIHR Biomedical Research Center (BRC), Metabolic Unit, Great Ormond Street Hospital and Institute of Child Health, University College London, London SE1 9RT, UK; [email protected] * Correspondence: [email protected]; Tel.: +48-606-415-888 Abstract: Most plasma proteins, cell membrane proteins and other proteins are glycoproteins with sugar chains attached to the polypeptide-glycans. Glycosylation is the main element of the post- translational transformation of most human proteins. Since glycosylation processes are necessary for many different biological processes, patients present a diverse spectrum of phenotypes and severity of symptoms. The most frequently observed neurological symptoms in congenital disorders of glycosylation (CDG) are: epilepsy, intellectual disability, myopathies, neuropathies and stroke-like episodes. Epilepsy is seen in many CDG subtypes and particularly present in the case of mutations -

Supplemental Material Placed on This Supplemental Material Which Has Been Supplied by the Author(S) J Med Genet
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) J Med Genet Supplement Supplementary Table S1: GENE MEAN GENE NAME OMIM SYMBOL COVERAGE CAKUT CAKUT ADTKD ADTKD aHUS/TMA aHUS/TMA TUBULOPATHIES TUBULOPATHIES Glomerulopathies Glomerulopathies Polycystic kidneys / Ciliopathies Ciliopathies / kidneys Polycystic METABOLIC DISORDERS AND OTHERS OTHERS AND DISORDERS METABOLIC x x ACE angiotensin-I converting enzyme 106180 139 x ACTN4 actinin-4 604638 119 x ADAMTS13 von Willebrand cleaving protease 604134 154 x ADCY10 adenylate cyclase 10 605205 81 x x AGT angiotensinogen 106150 157 x x AGTR1 angiotensin II receptor, type 1 106165 131 x AGXT alanine-glyoxylate aminotransferase 604285 173 x AHI1 Abelson helper integration site 1 608894 100 x ALG13 asparagine-linked glycosylation 13 300776 232 x x ALG9 alpha-1,2-mannosyltransferase 606941 165 centrosome and basal body associated x ALMS1 606844 132 protein 1 x x APOA1 apolipoprotein A-1 107680 55 x APOE lipoprotein glomerulopathy 107741 77 x APOL1 apolipoprotein L-1 603743 98 x x APRT adenine phosphoribosyltransferase 102600 165 x ARHGAP24 Rho GTPase-Activation protein 24 610586 215 x ARL13B ADP-ribosylation factor-like 13B 608922 195 x x ARL6 ADP-ribosylation factor-like 6 608845 215 ATPase, H+ transporting, lysosomal V0, x ATP6V0A4 605239 90 subunit a4 ATPase, H+ transporting, lysosomal x x ATP6V1B1 192132 163 56/58, V1, subunit B1 x ATXN10 ataxin -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Molecular Diagnostic Requisition
BAYLOR MIRACA GENETICS LABORATORIES SHIP TO: Baylor Miraca Genetics Laboratories 2450 Holcombe, Grand Blvd. -Receiving Dock PHONE: 800-411-GENE | FAX: 713-798-2787 | www.bmgl.com Houston, TX 77021-2024 Phone: 713-798-6555 MOLECULAR DIAGNOSTIC REQUISITION PATIENT INFORMATION SAMPLE INFORMATION NAME: DATE OF COLLECTION: / / LAST NAME FIRST NAME MI MM DD YY HOSPITAL#: ACCESSION#: DATE OF BIRTH: / / GENDER (Please select one): FEMALE MALE MM DD YY SAMPLE TYPE (Please select one): ETHNIC BACKGROUND (Select all that apply): UNKNOWN BLOOD AFRICAN AMERICAN CORD BLOOD ASIAN SKELETAL MUSCLE ASHKENAZIC JEWISH MUSCLE EUROPEAN CAUCASIAN -OR- DNA (Specify Source): HISPANIC NATIVE AMERICAN INDIAN PLACE PATIENT STICKER HERE OTHER JEWISH OTHER (Specify): OTHER (Please specify): REPORTING INFORMATION ADDITIONAL PROFESSIONAL REPORT RECIPIENTS PHYSICIAN: NAME: INSTITUTION: PHONE: FAX: PHONE: FAX: NAME: EMAIL (INTERNATIONAL CLIENT REQUIREMENT): PHONE: FAX: INDICATION FOR STUDY SYMPTOMATIC (Summarize below.): *FAMILIAL MUTATION/VARIANT ANALYSIS: COMPLETE ALL FIELDS BELOW AND ATTACH THE PROBAND'S REPORT. GENE NAME: ASYMPTOMATIC/POSITIVE FAMILY HISTORY: (ATTACH FAMILY HISTORY) MUTATION/UNCLASSIFIED VARIANT: RELATIONSHIP TO PROBAND: THIS INDIVIDUAL IS CURRENTLY: SYMPTOMATIC ASYMPTOMATIC *If family mutation is known, complete the FAMILIAL MUTATION/ VARIANT ANALYSIS section. NAME OF PROBAND: ASYMPTOMATIC/POPULATION SCREENING RELATIONSHIP TO PROBAND: OTHER (Specify clinical findings below): BMGL LAB#: A COPY OF ORIGINAL RESULTS ATTACHED IF PROBAND TESTING WAS PERFORMED AT ANOTHER LAB, CALL TO DISCUSS PRIOR TO SENDING SAMPLE. A POSITIVE CONTROL MAY BE REQUIRED IN SOME CASES. REQUIRED: NEW YORK STATE PHYSICIAN SIGNATURE OF CONSENT I certify that the patient specified above and/or their legal guardian has been informed of the benefits, risks, and limitations of the laboratory test(s) requested. -

Prenatal Testing Requisition Form
BAYLOR MIRACA GENETICS LABORATORIES SHIP TO: Baylor Miraca Genetics Laboratories 2450 Holcombe, Grand Blvd. -Receiving Dock PHONE: 800-411-GENE | FAX: 713-798-2787 | www.bmgl.com Houston, TX 77021-2024 Phone: 713-798-6555 PRENATAL COMPREHENSIVE REQUISITION FORM PATIENT INFORMATION NAME (LAST,FIRST, MI): DATE OF BIRTH (MM/DD/YY): HOSPITAL#: ACCESSION#: REPORTING INFORMATION ADDITIONAL PROFESSIONAL REPORT RECIPIENTS PHYSICIAN: NAME: INSTITUTION: PHONE: FAX: PHONE: FAX: NAME: EMAIL (INTERNATIONAL CLIENT REQUIREMENT): PHONE: FAX: SAMPLE INFORMATION CLINICAL INDICATION FETAL SPECIMEN TYPE Pregnancy at risk for specific genetic disorder DATE OF COLLECTION: (Complete FAMILIAL MUTATION information below) Amniotic Fluid: cc AMA PERFORMING PHYSICIAN: CVS: mg TA TC Abnormal Maternal Screen: Fetal Blood: cc GESTATIONAL AGE (GA) Calculation for AF-AFP* NTD TRI 21 TRI 18 Other: SELECT ONLY ONE: Abnormal NIPT (attach report): POC/Fetal Tissue, Type: TRI 21 TRI 13 TRI 18 Other: Cultured Amniocytes U/S DATE (MM/DD/YY): Abnormal U/S (SPECIFY): Cultured CVS GA ON U/S DATE: WKS DAYS PARENTAL BLOODS - REQUIRED FOR CMA -OR- Maternal Blood Date of Collection: Multiple Pregnancy Losses LMP DATE (MM/DD/YY): Parental Concern Paternal Blood Date of Collection: Other Indication (DETAIL AND ATTACH REPORT): *Important: U/S dating will be used if no selection is made. Name: Note: Results will differ depending on method checked. Last Name First Name U/S dating increases overall screening performance. Date of Birth: KNOWN FAMILIAL MUTATION/DISORDER SPECIFIC PRENATAL TESTING Notice: Prior to ordering testing for any of the disorders listed, you must call the lab and discuss the clinical history and sample requirements with a genetic counselor. -

Overview Gene List Target Scan Vs DIANA Group a Group B Group A
Overview Gene list Target scan vs DIANA Group A Group B Group A hsa-miR-181a hsa-miR-323 hsa-miR-326 Target scan Diana microT Overlap Target scan Diana microT Overlap Target scan SEPT3 SEPT3 SEPT3 SEPT7 ADARB1 HPCAL4 ABHD2 ABL2 ABHD13 ACVR2A ADCYAP1R1 AKAP13 PDPK1 ACRBP ACAN ABI1 ADAMTS1 ALAD APOBR ACVRL1 ACCN2 ABLIM1 ADAMTSL1 ANKRD52 ATXN1 ADAM19 ACER3 ACSL1 AKAP7 ARID2 C18orf23,RNF165 ADAM33 ACVR2A ACTN2 ANKRD43 ARL3 C20orf29 ADAMTS2 ADAMTS1 ACVR2A AP1S3 ARRB1 CACNG4 AHCYL2 ADAMTS18 ACVR2B ARID2 BBC3 CCNJL ALOX15B ADAMTS5 ADAM11 ATP11A BTG1 CYP2E1 ANK1 ADAMTSL1 ADAM22 ATXN1 C18orf62 GNB1L ANKS6 ADARB1 ADAMTS1 B4GALT1 C1orf21 GPR61 APBA1 AFAP1 ADAMTS6 BAG4 CADM4 GTSE1 ARCN1 AFTPH ADAMTSL1 BAI3 CALML4 HPCAL4 ARHGEF37 AK3 ADCY9 BNC2 CAPN6 KIAA0152 ARID3B AKAP7 ADRBK1 BRD1 CBFA2T2 KIF1A ARL8A ANAPC16 AFF2 BRWD1 CEBPA MACF1 ATP2B2 ANK1 AHCTF1,AHCTF1PBTBD3 CHD1 MYO1D ATP6V1G2 ANKRD12 AKAP2,PALM2 C13orf23 CIT PCNT AUP1 ANKRD33B AKAP6 C14orf43 CLASP2 PDPK1 BCL2L2 ANKRD43 AKAP7 CAPRIN1 CLCN5 PLEKHG4B BHLHE40 ANKRD44 AKAP9 CARM1 CLIP3 PPARA BTBD3 ANKRD52 AKT3 CBX4 COL5A2 PRB1,PRB2,PRB4 BTRC AP1S3 ALG9 CCDC117 CTNS PTPRT C10orf26 APBA1 ANKRD13C CCNJ DCTN4 PYCR1 C14orf1 APLP2 ANKRD20B CDH13 DCUN1D4 RAPGEF1 C16orf45 APOO ANKRD43 CDON DDB1 SRCAP C16orf54 ARID2 ANKRD50 CDYL DDX39B TMEM63C C1orf106 ARL3 AP1G1 CEP350 DIP2C C1orf27 ARRDC3 AP1S3 CHD7 DNAJB3 C22orf29 ATF7 API5 CHIC1 EEPD1 C9orf3 ATG2B ARFGEF2 CLIP1 EIF2C1 CACNA1E ATG7 ARHGAP12 CNOT6L ELFN2 CAPN12 ATP11A ARHGAP26 CNR1 ELK1 CASKIN1 ATP2B3 ARHGAP29 CNTN4 FAM172A CBFA2T3 ATP8B2 ARHGEF3 CNTNAP2 -

CDG-Ih) (ALG8 Deficiency
550 LETTER TO JMG J Med Genet: first published as 10.1136/jmg.2003.016923 on 2 July 2004. Downloaded from Clinical and molecular features of three patients with congenital disorders of glycosylation type Ih (CDG-Ih) (ALG8 deficiency) E Schollen, C G Frank, L Keldermans, R Reyntjens, C E Grubenmann, P T Clayton, B G Winchester, J Smeitink, R A Wevers, M Aebi, T Hennet, G Matthijs ............................................................................................................................... J Med Genet 2004;41:550–556. doi: 10.1136/jmg.2003.016923 rotein glycosylation is an essential post-translational Key points modification of various proteins, affecting their folding, Psorting, and function. Inborn defects in the assembly and processing of glycans on glycoproteins are known as N Congenital disorder of glycosylation type Ih (CDG-Ih) congenital disorders of glycosylation (CDG) and can affect is caused by a defect in the dolichyl-P-Glc:Glc1 both N- and O-glycosylation (for reviews see Marquardt and Man9GlcNAc2-PP-dolichyl a1,3-glucosyltransferase Denecke,1 Jaeken,2 and Grunewald et al3). N-glycosylation (ALG8). Thus far, the only patient has been described defects and especially defects in the assembly of the dolichol with a relatively mild clinical picture. linked N-glycan precursor in the endoplasmic reticulum (ER) N We describe the molecular and clinical features of (CDG-I) result in hypoglycosylation of many kinds of three patients from two families with an ALG8 (serum) glycoproteins. CDG-I is therefore a group of multi- deficiency. All three patients presented with severe, systemic disorders. life-threatening multi organ failure and died within The assembly of the N-glycan precursor in the ER is a their first months of life. -
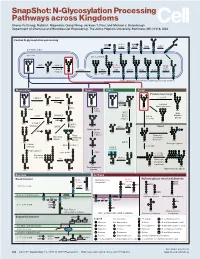
N-Glycosylation Processing Pathways Across Kingdoms Cheng-Yu Chung, Natalia I
SnapShot: N-Glycosylation Processing Pathways across Kingdoms Cheng-Yu Chung, Natalia I. Majewska, Qiong Wang, Jackson T. Paul, and Michael J. Betenbaugh Department of Chemical and Biomolecular Engineering, The Johns Hopkins University, Baltimore, MD 21218, USA Central N-glycosylation processing UDP ALG13 GDP ALG7 ALG14 ALG1 ALG2 ALG11 P P P P P CYTOPLASM P P P P P P GOLGI ER LUMEN GNTI α -Man IA En Bloc ALG3 Transfer ALG9 α-Man IB ER Man I α -Glc I OST ALG10 ALG8 ALG6 ALG12 Asn α -Man IC Asn Asn Asn α -Glc II P P P P P Asn P P P P P ALGs: asparagine-linked N-glycosylation processing enzymes Mammalian Insect Plant Fungi Filamentous fungi α-Man II FUT8 α-1,2-MT XYLT GDP Terminal Asn Asn Asn Asn UDP Asn Galactofuranose GNTII Asn Asn FUT8 f ± α-Gal Asn FUTC3 GNTII High FUT11 α -GALT B14GALTI Och1p Mannose FUT12 glycan α-Man II α-1,2-MT UDP Asn Asn ST3GAL4 Asn Asn GNTIV ST6GAL1 GNTIII GNTV Asn GNase Yeast Hybrid glycan P CMP GNase Asn Asn Tetra- α-Man II Mannosyl- Bisecting antennary phosphate glycan glycan transferase Asn Asn Asn Asn GALT1 Asn ST8SIA2 Core type glycan GNTs B14GALTI α-1,6-MT ST8SIA4 Asn Lewis A structure Polysialylation α-Man PolyLacNAc α-1,2-MT Alternative α-1,3-MT P sialic acid FUT13 ] [ STs Mannosyl- n phosphate Asn Asn Asn transferase Asn Asn Paucimannose Hypermannose glycan Asn Asn Asn glycans Bacteria Archaea Archaea glycan structural diversity Block transfer En Bloc Methanococcus En Bloc Transfer Transfer maripaludis OCH3 Thr OCH AgIB Thr OCH3 3 PERIPLASM PglB B B Asn P - P Asn OSO3 P Asn Asn INNER MEMBRANE P Asn P -

Identification of Genomic Targets of Krüppel-Like Factor 9 in Mouse Hippocampal
Identification of Genomic Targets of Krüppel-like Factor 9 in Mouse Hippocampal Neurons: Evidence for a role in modulating peripheral circadian clocks by Joseph R. Knoedler A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Neuroscience) in the University of Michigan 2016 Doctoral Committee: Professor Robert J. Denver, Chair Professor Daniel Goldman Professor Diane Robins Professor Audrey Seasholtz Associate Professor Bing Ye ©Joseph R. Knoedler All Rights Reserved 2016 To my parents, who never once questioned my decision to become the other kind of doctor, And to Lucy, who has pushed me to be a better person from day one. ii Acknowledgements I have a huge number of people to thank for having made it to this point, so in no particular order: -I would like to thank my adviser, Dr. Robert J. Denver, for his guidance, encouragement, and patience over the last seven years; his mentorship has been indispensable for my growth as a scientist -I would also like to thank my committee members, Drs. Audrey Seasholtz, Dan Goldman, Diane Robins and Bing Ye, for their constructive feedback and their willingness to meet in a frequently cold, windowless room across campus from where they work -I am hugely indebted to Pia Bagamasbad and Yasuhiro Kyono for teaching me almost everything I know about molecular biology and bioinformatics, and to Arasakumar Subramani for his tireless work during the home stretch to my dissertation -I am grateful for the Neuroscience Program leadership and staff, in particular -

Ordered Assembly of the Asymmetrically Branched Lipid
CORE Metadata, citation and similar papers at core.ac.uk Provided by REROGlycobiology DOC Digital vol. Library 9 no. 6 pp. 617–625, 1999 Ordered assembly of the asymmetrically branched lipid-linked oligosaccharide in the endoplasmic reticulum is ensured by the substrate specificity of the individual glycosyltransferases Patricie Burda, Claude A.Jakob1, Jens Beinhauer2, identified (Herscovics and Orlean, 1993; Orlean, 1997; Burda Johannes H.Hegemann2 and Markus Aebi3 and Aebi, 1999). A common characteristic of the different alg Mikrobiologisches Institut, ETH Zürich, CH-8092 Zürich, Switzerland, mutant strains is the accumulation of a biosynthetic oligosacchar- 1Division of Cell and Molecular Pathology, Department of Pathology, ide intermediate specific for the defective ALG locus. Based on University of Zurich, CH-8091 Zurich, Switzerland and the assumption that the accumulating intermediate is the acceptor 2Heinrich-Heine-Universität Düsseldorf, Institut für Mikrobiologie, D-40225 oligosaccharide of the reaction affected, defined glycosyltrans- Düsseldorf, Germany ferase activities could be assigned to the different ALG loci Received on September 15, 1998; revised on October 16, 1998; accepted on (Burda and Aebi, 1999). In addition, mutations in ALG loci lead October 27, 1998 to underglycosylation of secreted proteins in vivo. The reason for this underglycosylation appears to be the decreased affinity of the 3To whom correspondence should be addressed at: Mikrobiologisches Institut, ETH Zentrum, LFV E20, 8092 Zürich, Switzerland oligosaccharyltransferase