Properties of Vinorine Synthase — the Rauwolfia Enzyme Involved in the Formation of the Ajmaline Skeleton
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Download Article (PDF)
Enzymatic Biosynthesis of Vomilenine, a Key Intermediate of the Ajmaline Pathway, Catalyzed by a Novel Cytochrome P 450-Dependent Enzyme from Plant Cell Cultures of Rauwolfia serpentina Heike Falkenhagen and Joachim Stöckigt Lehrstuhl für Pharmazeutische Biologie der Johannes Gutenberg-Universität Mainz, Institut für Pharmazie, Staudinger Weg 5, D-55099 Mainz, Bundesrepublik Deutschland Z. Naturforsch. 50c, 45-53 (1995); received September 26, 1994 Vomilenine, Vinorine, Vinorine Hydroxylase, Cytochrome P450, Rauwolfia serpentina Microsomal preparations from Rauwolfia serpentina Benth. cell suspension cultures cata lyze a key step in the biosynthesis of ajmaline - the enzymatic hydroxylation of the indole alkaloid vinorine at the allylic C-21 resulting in vomilenine. Vomilenine is an important branch-point intermediate, leading not only to ajmaline but also to several side reactions of the biosynthetic pathway to ajmaline. The investigation of the taxonomical distribution of the enzyme indicated that vinorine hydroxylase is exclusively present in ajmaline-producing plant cells. The novel enzyme is strictly dependent on NADPH2 and 0 2 and can be inhibited by typical cytochrome P450 inhibitors such as cytochrome c, ketoconazole and carbon mon oxide (the effect of CO is reversible with light of 450 nm). This suggests that vinorine hy droxylase is a cytochrome P450-dependent monooxygenase. A pH optimum of 8.3 and a temperature optimum of 40 °C were found. The Km value was 3 for NADPH2 and 26 [i,M for vinorine. The optimum enzyme activity could be determined at day 4 after inoculation of the cell cultures in AP I medium. Vinorine hydroxylase could be stored with 20% sucrose at -28 °C without significant loss of activity. -

Evolutionary Routes to Biochemical Innovation Revealed by Integrative
RESEARCH ARTICLE Evolutionary routes to biochemical innovation revealed by integrative analysis of a plant-defense related specialized metabolic pathway Gaurav D Moghe1†, Bryan J Leong1,2, Steven M Hurney1,3, A Daniel Jones1,3, Robert L Last1,2* 1Department of Biochemistry and Molecular Biology, Michigan State University, East Lansing, United States; 2Department of Plant Biology, Michigan State University, East Lansing, United States; 3Department of Chemistry, Michigan State University, East Lansing, United States Abstract The diversity of life on Earth is a result of continual innovations in molecular networks influencing morphology and physiology. Plant specialized metabolism produces hundreds of thousands of compounds, offering striking examples of these innovations. To understand how this novelty is generated, we investigated the evolution of the Solanaceae family-specific, trichome- localized acylsugar biosynthetic pathway using a combination of mass spectrometry, RNA-seq, enzyme assays, RNAi and phylogenomics in different non-model species. Our results reveal hundreds of acylsugars produced across the Solanaceae family and even within a single plant, built on simple sugar cores. The relatively short biosynthetic pathway experienced repeated cycles of *For correspondence: [email protected] innovation over the last 100 million years that include gene duplication and divergence, gene loss, evolution of substrate preference and promiscuity. This study provides mechanistic insights into the † Present address: Section of emergence of plant chemical novelty, and offers a template for investigating the ~300,000 non- Plant Biology, School of model plant species that remain underexplored. Integrative Plant Sciences, DOI: https://doi.org/10.7554/eLife.28468.001 Cornell University, Ithaca, United States Competing interests: The authors declare that no Introduction competing interests exist. -

3D-Structure of Vinorine Synthase from Rauvolfia Serpentina in Complex with Its Ligand Acetyl-Coa
3D-Structure of Vinorine Synthase from Rauvolfia serpentina in Complex with its Ligand Acetyl-CoA M. Hill, S. Panjikar1, X. Ma, J. Stöckigt Department of Pharmaceutical Biology, Institute of Pharmacy, Johannes Gutenberg-University Mainz, Staudingerweg 5, D-55099 Mainz, Germany 1EMBL Hamburg outstation DESY, Notkestraße 85, D-22607 Hamburg, Germany Vinorine synthase (VS, EC 2.3.1.160) is an acetyl transferase that occupies a central role in the biosynthesis of the antiarrhythmic monoterpenoid indole alkaloid ajmaline in the Indian medicinal plant Rauvolfia serpentina. The enzyme catalyzes the reversible acetyl-CoA dependent formation of the ajmalan-type alkaloid vinorine from the sarpagine-type alkaloid 16-epi-vellosimine (Fig. 1) [1]. VS belongs to the BAHD (benzylalcohol acetyl-, anthocyanin-O-hydroxy-cinnamoyl, anthranilate-N-hydroxy-cinnamoyl/benzoyl-, deacetylvindoline acetyltransferase) enzyme superfamily of acetyl transferases, which members are involved in biosynthesis of several important drugs (morphine, taxol, vincaleucoblastine and vincristine) and other products of secondary metabolism in plants. The synthase consists of 412 amino acids and has a molecular weight of 46.8 kDa. All conserved residues typical for the BAHD family are found in VS. Prominent features of this family are the HxxxD motif in the active site and the DFGWG motif near the C-terminus. The typical low overall sequence identity (25-34%) to other BAHD members might indicate a divergent evolution of the family from one ancestral gene [2]. O H C H OAc 16 AcCoA CoA N N N H H VS N H AcCoA CoA 16-epi- Vinorin Figure 1: Reaction catalyzed by vinorine synthase (VS) Vinorine synthase from R. -

Identification and Characterization of Piperine Synthase from Black
ARTICLE https://doi.org/10.1038/s42003-021-01967-9 OPEN Identification and characterization of piperine synthase from black pepper, Piper nigrum L. Arianne Schnabel 1, Benedikt Athmer1, Kerstin Manke1, Frank Schumacher2, Fernando Cotinguiba 3 & ✉ Thomas Vogt 1 Black pepper (Piper nigrum L.) is the world’s most popular spice and is also used as an ingredient in traditional medicine. Its pungent perception is due to the interaction of its major compound, piperine (1-piperoyl-piperidine) with the human TRPV-1 or vanilloid receptor. We now identify the hitherto concealed enzymatic formation of piperine from piperoyl coenzyme A and piperidine based on a differential RNA-Seq approach from developing black pepper 1234567890():,; fruits. This enzyme is described as piperine synthase (piperoyl-CoA:piperidine piperoyl transferase) and is a member of the BAHD-type of acyltransferases encoded by a gene that is preferentially expressed in immature fruits. A second BAHD-type enzyme, also highly expressed in immature black pepper fruits, has a rather promiscuous substrate specificity, combining diverse CoA-esters with aliphatic and aromatic amines with similar efficiencies, and was termed piperamide synthase. Recombinant piperine and piperamide synthases are members of a small gene family in black pepper. They can be used to facilitate the microbial production of a broad range of medicinally relevant aliphatic and aromatic piperamides based on a wide array of CoA-donors and amine-derived acceptors, offering widespread applications. 1 Leibniz Institute of Plant Biochemistry, Dept. Cell and Metabolic Biology, Halle (Saale), Germany. 2 Core Facility Vienna Botanical Gardens, Vienna, Austria. ✉ 3 Instituto de Pesquisas de Produtos Naturais (IPPN), Universidade Federal do Rio de Janeiro (UFRJ), Rio de Janeiro/RJ, Brasil. -
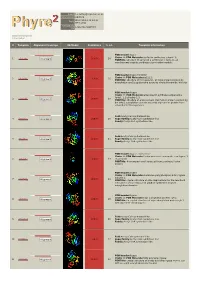
Phyre 2 Results for Q9VDC6
Email [email protected] Description Q9VDC6 Wed Feb 13 11:14:11 Date GMT 2013 Unique Job c7a32c1e7988f754 ID Detailed template information # Template Alignment Coverage 3D Model Confidence % i.d. Template Information PDB header:ligase Chain: A: PDB Molecule:surfactin synthetase subunit 3; 1 c2vsqA_ 100.0 28 Alignment PDBTitle: structure of surfactin a synthetase c (srfa-c), a2 nonribosomal peptide synthetase termination module PDB header:ligase/inhibitor Chain: A: PDB Molecule:pa1221; 2 c4dg9A_ 100.0 25 Alignment PDBTitle: structure of holo-pa1221, an nrps protein containing adenylation and2 pcp domains bound to vinylsulfonamide inhibitor PDB header:ligase Chain: H: PDB Molecule:enterobactin synthase component e (ente), 2,3-dihydro-2,3- 3 c3rg2H_ 100.0 19 Alignment PDBTitle: structure of a two-domain nrps fusion protein containing the ente2 adenylation domain and entb aryl-carrier protein from enterobactin3 biosynthesis Fold:Acetyl-CoA synthetase-like 4 d1pg4a_ Alignment 100.0 20 Superfamily:Acetyl-CoA synthetase-like Family:Acetyl-CoA synthetase-like Fold:Acetyl-CoA synthetase-like 5 d1ry2a_ Alignment 100.0 21 Superfamily:Acetyl-CoA synthetase-like Family:Acetyl-CoA synthetase-like PDB header:ligase, transferase Chain: A: PDB Molecule:fusion protein 4-coumarate--coa ligase 1, 6 c3tsyA_ Alignment 100.0 17 resveratrol PDBTitle: 4-coumaroyl-coa ligase::stilbene synthase fusion protein PDB header:ligase Chain: A: PDB Molecule:d-alanine--poly(phosphoribitol) ligase subunit 1; 7 c3e7wA_ 100.0 23 Alignment PDBTitle: crystal structure -

Solution of the Multistep Pathway for Assembly of Corynanthean, Strychnos, Iboga, and Aspidosperma Monoterpenoid Indole Alkaloids from 19E-Geissoschizine
Solution of the multistep pathway for assembly of corynanthean, strychnos, iboga, and aspidosperma monoterpenoid indole alkaloids from 19E-geissoschizine Yang Qua, Michael E. A. M. Eassona,1, Razvan Simionescub, Josef Hajicekb,2, Antje M. K. Thamma,3, Vonny Salima,4, and Vincenzo De Lucaa,5 aDepartment of Biological Sciences, Brock University, St. Catharines, ON L2S 3A1, Canada; and bDepartment of Chemistry, Brock University, St. Catharines, ON L2S 3A1, Canada Edited by Jerrold Meinwald, Cornell University, Ithaca, NY, and approved February 11, 2018 (received for review November 16, 2017) Monoterpenoid indole alkaloids (MIAs) possess a diversity of for the formation of tabersonine or catharanthine from 19E- alkaloid skeletons whose biosynthesis is poorly understood. A geissoschizine. Gene discovery involved a combination of bio- bioinformatic search of candidate genes, combined with their informatics and virus-induced gene silencing (VIGS) to identify virus-induced gene silencing, targeted MIA profiling and in vitro/ candidate genes and functional expression of the selected genes in vivo pathway reconstitution identified and functionally charac- by in vitro/in vivo reconstitution of the pathway. A series of terized six genes as well as a seventh enzyme reaction required for highly unstable intermediates that rearrange to other important the conversion of 19E-geissoschizine to tabersonine and catharan- MIA precursors when not used by appropriate enzymes reveals thine. The involvement of pathway intermediates in the formation the plasticity of MIA formation and how this fundamental of four MIA skeletons is described, and the role of stemmadenine- property led to diverse MIA structures found in nature. O-acetylation in providing necessary reactive substrates for the formation of iboga and aspidosperma MIAs is described. -

12) United States Patent (10
US007635572B2 (12) UnitedO States Patent (10) Patent No.: US 7,635,572 B2 Zhou et al. (45) Date of Patent: Dec. 22, 2009 (54) METHODS FOR CONDUCTING ASSAYS FOR 5,506,121 A 4/1996 Skerra et al. ENZYME ACTIVITY ON PROTEIN 5,510,270 A 4/1996 Fodor et al. MICROARRAYS 5,512,492 A 4/1996 Herron et al. 5,516,635 A 5/1996 Ekins et al. (75) Inventors: Fang X. Zhou, New Haven, CT (US); 5,532,128 A 7/1996 Eggers Barry Schweitzer, Cheshire, CT (US) 5,538,897 A 7/1996 Yates, III et al. s s 5,541,070 A 7/1996 Kauvar (73) Assignee: Life Technologies Corporation, .. S.E. al Carlsbad, CA (US) 5,585,069 A 12/1996 Zanzucchi et al. 5,585,639 A 12/1996 Dorsel et al. (*) Notice: Subject to any disclaimer, the term of this 5,593,838 A 1/1997 Zanzucchi et al. patent is extended or adjusted under 35 5,605,662 A 2f1997 Heller et al. U.S.C. 154(b) by 0 days. 5,620,850 A 4/1997 Bamdad et al. 5,624,711 A 4/1997 Sundberg et al. (21) Appl. No.: 10/865,431 5,627,369 A 5/1997 Vestal et al. 5,629,213 A 5/1997 Kornguth et al. (22) Filed: Jun. 9, 2004 (Continued) (65) Prior Publication Data FOREIGN PATENT DOCUMENTS US 2005/O118665 A1 Jun. 2, 2005 EP 596421 10, 1993 EP 0619321 12/1994 (51) Int. Cl. EP O664452 7, 1995 CI2O 1/50 (2006.01) EP O818467 1, 1998 (52) U.S. -

Integrative Itraq-Based Proteomic and Transcriptomic Analysis Reveals
Ye et al. Horticulture Research (2021) 8:157 Horticulture Research https://doi.org/10.1038/s41438-021-00591-2 www.nature.com/hortres ARTICLE Open Access Integrative iTRAQ-based proteomic and transcriptomic analysis reveals the accumulation patterns of key metabolites associated with oil quality during seed ripening of Camellia oleifera Zhouchen Ye1,JingYu1,WupingYan1,JunfengZhang1, Dongmei Yang1, Guanglong Yao1, Zijin Liu1, Yougen Wu 1 and Xilin Hou 2 Abstract Camellia oleifera (C. oleifera) is one of the four major woody oil-bearing crops in the world and has relatively high ecological, economic, and medicinal value. Its seeds undergo a series of complex physiological and biochemical changes during ripening, which is mainly manifested as the accumulation and transformation of certain metabolites closely related to oil quality, especially flavonoids and fatty acids. To obtain new insights into the underlying molecular mechanisms, a parallel analysis of the transcriptome and proteome profiles of C. oleifera seeds at different maturity levels was conducted using RNA sequencing (RNA-seq) and isobaric tags for relative and absolute quantification (iTRAQ) complemented with gas chromatography-mass spectrometry (GC-MS) data. A total of 16,530 transcripts and 1228 proteins were recognized with significant differential abundances in pairwise comparisons of samples at various developmental stages. Among these, 317 were coexpressed with a poor correlation, and most were involved in metabolic processes, including fatty acid metabolism, α-linolenic acid metabolism, and glutathione metabolism. In addition, the content of total flavonoids decreased gradually 1234567890():,; 1234567890():,; 1234567890():,; 1234567890():,; with seed maturity, and the levels of fatty acids generally peaked at the fat accumulation stage; these results basically agreed with the regulation patterns of genes or proteins in the corresponding pathways. -

Structural and Functional Characterization of The
Supplemental Material can be found at: http://www.jbc.org/cgi/content/full/M705752200/DC1 THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 283, NO. 3, pp. 1660–1669, January 18, 2008 Printed in the U.S.A. Structural and Functional Characterization of the TRI101 Trichothecene 3-O-Acetyltransferase from Fusarium sporotrichioides and Fusarium graminearum KINETIC INSIGHTS TO COMBATING FUSARIUM HEAD BLIGHT*□S Received for publication, July 13, 2007, and in revised form, October 1, 2007 Published, JBC Papers in Press, October 8, 2007, DOI 10.1074/jbc.M705752200 Graeme S. Garvey‡, Susan P. McCormick§, and Ivan Rayment‡1 From the ‡Department of Biochemistry, University of Wisconsin, Madison, Wisconsin 53706 and the §Mycotoxin Research Unit, USDA/ARS, National Center for Agricultural Utilization Research, Peoria, Illinois 61604 Downloaded from Fusarium head blight (FHB) is a plant disease with serious Fusarium head blight (FHB)2 is a plant disease with serious economic and health impacts. It is caused by fungal species economic and health consequences (1, 2). The disease is caused belonging to the genus Fusarium and the mycotoxins they by several species of the fungus Fusarium that reduce grain produce. Although it has proved difficult to combat this dis- yields and contaminate the grains with trichothecene mycotox- ease, one strategy that has been examined is the introduction ins such as deoxynivalenol, nivalenol, and T-2 toxin. In the www.jbc.org of an indigenous fungal protective gene into cereals such as United States and Europe, deoxynivalenol (DON) and wheat barley and rice. Thus far the gene of choice has been 15-acetyl-DON are the primary grain contaminants, where tri101 whose gene product catalyzes the transfer of an acetyl these are produced by strains of Fusarium graminearum (3, 4). -

Enzymatic Biosynthesis of Vomilenine, a Key Intermediate Of
Enzymatic Biosynthesis of Vomilenine, a Key Intermediate of the Ajmaline Pathway, Catalyzed by a Novel Cytochrome P 450-Dependent Enzyme from Plant Cell Cultures of Rauwolfia serpentina Heike Falkenhagen and Joachim Stöckigt Lehrstuhl für Pharmazeutische Biologie der Johannes Gutenberg-Universität Mainz, Institut für Pharmazie, Staudinger Weg 5, D-55099 Mainz, Bundesrepublik Deutschland Z. Naturforsch. 50c, 45-53 (1995); received September 26, 1994 Vomilenine, Vinorine, Vinorine Hydroxylase, Cytochrome P450, Rauwolfia serpentina Microsomal preparations from Rauwolfia serpentina Benth. cell suspension cultures cata lyze a key step in the biosynthesis of ajmaline - the enzymatic hydroxylation of the indole alkaloid vinorine at the allylic C-21 resulting in vomilenine. Vomilenine is an important branch-point intermediate, leading not only to ajmaline but also to several side reactions of the biosynthetic pathway to ajmaline. The investigation of the taxonomical distribution of the enzyme indicated that vinorine hydroxylase is exclusively present in ajmaline-producing plant cells. The novel enzyme is strictly dependent on NADPH2 and 0 2 and can be inhibited by typical cytochrome P450 inhibitors such as cytochrome c, ketoconazole and carbon mon oxide (the effect of CO is reversible with light of 450 nm). This suggests that vinorine hy droxylase is a cytochrome P450-dependent monooxygenase. A pH optimum of 8.3 and a temperature optimum of 40 °C were found. The Km value was 3 for NADPH2 and 26 [i,M for vinorine. The optimum enzyme activity could be determined at day 4 after inoculation of the cell cultures in AP I medium. Vinorine hydroxylase could be stored with 20% sucrose at -28 °C without significant loss of activity. -

Systemin Involvement in Tomato Defense Priming
SYSTEMIN INVOLVEMENT IN TOMATO DEFENSE PRIMING Valentina Madonna Dottorato in Scienze Biotecnologiche – XXVIII ciclo Indirizzo Biotecnologie per le Produzioni Vegetali Università di Napoli Federico II Dottorato in Scienze Biotecnologiche – XXVIII ciclo Indirizzo Biotecnologie per le Produzioni Vegetali Università di Napoli Federico II SYSTEMIN INVOLVEMENT IN TOMATO DEFENSE PRIMING Valentina Madonna Dottoranda: Valentina Madonna Relatore: Prof.ssa Rosa Rao Coordinatore: Prof. Giovanni Sannia A Francesco e alla mia Famiglia INDEX RIASSUNTO ........................................................................................................... 1 SUMMARY ............................................................................................................. 7 1. INTRODUCTION ................................................................................................ 9 1.1 Plant undergo a series of different stresses ................................................... 9 1.2 Plant defenses against biotic stresses: Direct and Indirect .......................... 10 1.3 Time of defenses: Constitutive and Inducible responses ............................. 12 1.4 Plant responses against insects .................................................................. 14 1.5 Priming of defences ..................................................................................... 16 1.6 Solanum lycopersicum defence: Systemin .................................................. 19 1.7 Octadecanoid pathway ............................................................................... -

Rough Draft of Dissertation
Tropane alkaloid biosynthesis in Erythroxylum coca involves an atypical type III polyketide synthase by Neill Kim, B.A. A Dissertation In Chemistry Submitted to the Graduate Faculty of Texas Tech University in Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY Approved Dr. Michael Latham Chair of Committee Dr. John D’Auria Dr. Joachim Weber Mark Sheridan Dean of the Graduate School May, 2020 Copyright 2020, Neill Kim Texas Tech University, Neill Kim, May 2020 ACKNOWLEDGMENTS I would like to thank Texas Tech University for the resources and support they provided, Dr. John D’Auria for all the guidance and support he has given me, and Dr. Michael Latham. I would also like the thank Dr. Charles Stewart for helping with the crystallography of the enzyme. This research was funded by the National Science Foundation under grant No. NSF-171423326 given to Dr. John D’Auria. ii Texas Tech University, Neill Kim, May 2020 TABLE OF CONTENTS ACKNOWLEDGMENTS ........................................................................................... ii ABSTRACT ................................................................................................................. vi LIST OF TABLES ..................................................................................................... vii LIST OF FIGURES .................................................................................................. viii LIST OF SCHEMES ................................................................................................