Download File
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Vocalization Behavior of the Endangered Bahama Oriole (Icterus Northropi): Ontogenetic, Sexual, Temporal, Duetting Pair, and Geographic Variation Valerie A
Loma Linda University TheScholarsRepository@LLU: Digital Archive of Research, Scholarship & Creative Works Loma Linda University Electronic Theses, Dissertations & Projects 3-1-2011 Vocalization Behavior of the Endangered Bahama Oriole (Icterus northropi): Ontogenetic, Sexual, Temporal, Duetting Pair, and Geographic Variation Valerie A. Lee Loma Linda University Follow this and additional works at: http://scholarsrepository.llu.edu/etd Part of the Biology Commons Recommended Citation Lee, Valerie A., "Vocalization Behavior of the Endangered Bahama Oriole (Icterus northropi): Ontogenetic, Sexual, Temporal, Duetting Pair, and Geographic Variation" (2011). Loma Linda University Electronic Theses, Dissertations & Projects. 37. http://scholarsrepository.llu.edu/etd/37 This Thesis is brought to you for free and open access by TheScholarsRepository@LLU: Digital Archive of Research, Scholarship & Creative Works. It has been accepted for inclusion in Loma Linda University Electronic Theses, Dissertations & Projects by an authorized administrator of TheScholarsRepository@LLU: Digital Archive of Research, Scholarship & Creative Works. For more information, please contact [email protected]. LOMA LINDA UNIVERSITY School of Science and Technology in conjunction with the Faculty of Graduate Studies ____________________ Vocalization Behavior of the Endangered Bahama Oriole (Icterus northropi): Ontogenetic, Sexual, Temporal, Duetting Pair, and Geographic Variation by Valerie A. Lee ____________________ A Thesis submitted in partial satisfaction of the requirements for the degree of Master of Science in Biology ____________________ March 2011 © 2011 Valerie A. Lee All Rights Reserved Each person whose signature appears below certifies that this thesis in his/her opinion is adequate, in scope and quality, as a thesis for the degree Master of Science. , Chairperson William K. Hayes, Professor of Biology Stephen G. -

Song and Plumage Evolution in the New World Orioles (Icterus) Show Similar Lability and Convergence in Patterns
ORIGINAL ARTICLE doi:10.1111/j.1558-5646.2007.00082.x SONG AND PLUMAGE EVOLUTION IN THE NEW WORLD ORIOLES (ICTERUS) SHOW SIMILAR LABILITY AND CONVERGENCE IN PATTERNS J. Jordan Price,1,2 Nicholas R. Friedman,1,3 and Kevin E. Omland4,5 1Department of Biology, St. Mary’s College of Maryland, St. Mary’s City, Maryland 20686 2E-mail: [email protected] 3E-mail: [email protected] 4Department of Biological Sciences, University of Maryland, Baltimore County, Baltimore, Maryland 21250 5E-mail: [email protected] Received August 28, 2006 Accepted November 23, 2006 Both song and color patterns in birds are thought to evolve rapidly and exhibit high levels of homoplasy, yet few previous studies have compared the evolution of these traits systematically using the same taxa. Here we reconstruct the evolution of song in the New World orioles (Icterus) and compare patterns of vocal evolution to previously reconstructed patterns of change in plumage evolution in this clade. Individual vocal characters exhibit high levels of homoplasy, reflected in a low overall consistency index (CI = 0.27) and retention index (RI = 0.35). Levels of lability in song are comparable to those found for oriole plumage patterns using the same taxa (CI = 0.31, RI = 0.63), but are strikingly dissimilar to the conservative patterns of change seen in the songs of oropendolas (Psarocolius, Ocyalus;CI= 0.82, RI = 0.87), a group closely related to the orioles. Oriole song is also similar to oriole plumage in exhibiting repeated convergence in overall patterns, with some distantly related taxa sounding remarkably similar. -

Environmental Influences on Endothelial Gene Expression
ENDOTHELIAL CELL GENE EXPRESSION John Matthew Jeff Herbert Supervisors: Prof. Roy Bicknell and Dr. Victoria Heath PhD thesis University of Birmingham August 2012 University of Birmingham Research Archive e-theses repository This unpublished thesis/dissertation is copyright of the author and/or third parties. The intellectual property rights of the author or third parties in respect of this work are as defined by The Copyright Designs and Patents Act 1988 or as modified by any successor legislation. Any use made of information contained in this thesis/dissertation must be in accordance with that legislation and must be properly acknowledged. Further distribution or reproduction in any format is prohibited without the permission of the copyright holder. ABSTRACT Tumour angiogenesis is a vital process in the pathology of tumour development and metastasis. Targeting markers of tumour endothelium provide a means of targeted destruction of a tumours oxygen and nutrient supply via destruction of tumour vasculature, which in turn ultimately leads to beneficial consequences to patients. Although current anti -angiogenic and vascular targeting strategies help patients, more potently in combination with chemo therapy, there is still a need for more tumour endothelial marker discoveries as current treatments have cardiovascular and other side effects. For the first time, the analyses of in-vivo biotinylation of an embryonic system is performed to obtain putative vascular targets. Also for the first time, deep sequencing is applied to freshly isolated tumour and normal endothelial cells from lung, colon and bladder tissues for the identification of pan-vascular-targets. Integration of the proteomic, deep sequencing, public cDNA libraries and microarrays, delivers 5,892 putative vascular targets to the science community. -

A Comprehensive Species-Level Molecular Phylogeny of the New World
YMPEV 4758 No. of Pages 19, Model 5G 2 December 2013 Molecular Phylogenetics and Evolution xxx (2013) xxx–xxx 1 Contents lists available at ScienceDirect Molecular Phylogenetics and Evolution journal homepage: www.elsevier.com/locate/ympev 5 6 3 A comprehensive species-level molecular phylogeny of the New World 4 blackbirds (Icteridae) a,⇑ a a b c d 7 Q1 Alexis F.L.A. Powell , F. Keith Barker , Scott M. Lanyon , Kevin J. Burns , John Klicka , Irby J. Lovette 8 a Department of Ecology, Evolution and Behavior, and Bell Museum of Natural History, University of Minnesota, 100 Ecology Building, 1987 Upper Buford Circle, St. Paul, MN 9 55108, USA 10 b Department of Biology, San Diego State University, San Diego, CA 92182, USA 11 c Barrick Museum of Natural History, University of Nevada, Las Vegas, NV 89154, USA 12 d Fuller Evolutionary Biology Program, Cornell Lab of Ornithology, Cornell University, 159 Sapsucker Woods Road, Ithaca, NY 14950, USA 1314 15 article info abstract 3117 18 Article history: The New World blackbirds (Icteridae) are among the best known songbirds, serving as a model clade in 32 19 Received 5 June 2013 comparative studies of morphological, ecological, and behavioral trait evolution. Despite wide interest in 33 20 Revised 11 November 2013 the group, as yet no analysis of blackbird relationships has achieved comprehensive species-level sam- 34 21 Accepted 18 November 2013 pling or found robust support for most intergeneric relationships. Using mitochondrial gene sequences 35 22 Available online xxxx from all 108 currently recognized species and six additional distinct lineages, together with strategic 36 sampling of four nuclear loci and whole mitochondrial genomes, we were able to resolve most relation- 37 23 Keywords: ships with high confidence. -

21 Sep 2018 Lists of Victims and Hosts of the Parasitic
version: 21 Sep 2018 Lists of victims and hosts of the parasitic cowbirds (Molothrus). Peter E. Lowther, Field Museum Brood parasitism is an awkward term to describe an interaction between two species in which, as in predator-prey relationships, one species gains at the expense of the other. Brood parasites "prey" upon parental care. Victimized species usually have reduced breeding success, partly because of the additional cost of caring for alien eggs and young, and partly because of the behavior of brood parasites (both adults and young) which may directly and adversely affect the survival of the victim's own eggs or young. About 1% of all bird species, among 7 families, are brood parasites. The 5 species of brood parasitic “cowbirds” are currently all treated as members of the genus Molothrus. Host selection is an active process. Not all species co-occurring with brood parasites are equally likely to be selected nor are they of equal quality as hosts. Rather, to varying degrees, brood parasites are specialized for certain categories of hosts. Brood parasites may rely on a single host species to rear their young or may distribute their eggs among many species, seemingly without regard to any characteristics of potential hosts. Lists of species are not the best means to describe interactions between a brood parasitic species and its hosts. Such lists do not necessarily reflect the taxonomy used by the brood parasites themselves nor do they accurately reflect the complex interactions within bird communities (see Ortega 1998: 183-184). Host lists do, however, offer some insight into the process of host selection and do emphasize the wide variety of features than can impact on host selection. -

Advances in Prognostic Methylation Biomarkers for Prostate Cancer
cancers Review Advances in Prognostic Methylation Biomarkers for Prostate Cancer 1 1,2 1,2, 1,2, , Dilys Lam , Susan Clark , Clare Stirzaker y and Ruth Pidsley * y 1 Epigenetics Research Laboratory, Genomics and Epigenetics Division, Garvan Institute of Medical Research, Sydney, New South Wales 2010, Australia; [email protected] (D.L.); [email protected] (S.C.); [email protected] (C.S.) 2 St. Vincent’s Clinical School, University of New South Wales, Sydney, New South Wales 2010, Australia * Correspondence: [email protected]; Tel.: +61-2-92958315 These authors have contributed equally. y Received: 22 September 2020; Accepted: 13 October 2020; Published: 15 October 2020 Simple Summary: Prostate cancer is a major cause of cancer-related death in men worldwide. There is an urgent clinical need for improved prognostic biomarkers to better predict the likely outcome and course of the disease and thus inform the clinical management of these patients. Currently, clinically recognised prognostic markers lack sensitivity and specificity in distinguishing aggressive from indolent disease, particularly in patients with localised, intermediate grade prostate cancer. Thus, there is major interest in identifying new molecular biomarkers to complement existing standard clinicopathological markers. DNA methylation is a frequent alteration in the cancer genome and offers potential as a reliable and robust biomarker. In this review, we provide a comprehensive overview of the current state of DNA methylation biomarker studies in prostate cancer prognosis. We highlight advances in this field that have enabled the discovery of novel prognostic genes and discuss the potential of methylation biomarkers for noninvasive liquid-biopsy testing. -

Supplemental Wing Shape and Dispersal Analysis
Data Supplement High dispersal ability inhibits speciation in a continental radiation of passerine birds Santiago Claramunt, Elizabeth P. Derryberry, J. V. Remsen, Jr. & Robb T. Brumfield Museum of Natural Science and Department of Biological Sciences, Louisiana State University, Baton Rouge, LA 70803, USA HAND-WING INDEX AND FLIGHT PERFORMANCE IN NEOTROPICAL FOREST BIRDS We investigated the relationship between wing shape and flight distances determined during 'dispersal challenge' experiments conducted in Gatun Lake in the Panama Canal (Moore et al. 2008). During the experiments, birds were released from a boat at incremental distances from shore and the distance flown or the success or failure in reaching the coast was recorded. To investigated the relationship between the hand-wing index and flight distance in Neotropical birds we used data on mean distance flown from table 3 in ref. We estimated hand-wing indices for the 10 species reported in those experiments (Table S1) . Wing measurements were taken by SC for four males of each species at LSUMNS. The relationship between the hand-wing index and distance flown was evaluated statistically using phylogenetic generalized least-squares (PGLS, Freckleton et al. 2002). We generated a phylogeny for the species involved in the experiment or an appropriate surrogate using DNA sequences of the slow-evolving RAG 1 gene from GenBank (Table S2). A maximum likelihood ultrametric tree was generated in PAUP* (Swofford 2003) using a GTR+! model of nucleotide substitution rates, empirical nucleotide frequencies, and enforcing a molecular clock. We found that the hand-wing index was strongly related to mean distance flown (R2 = 0.68, F = 20, d.f. -

Evolution of the Ovenbird-Woodcreeper Assemblage (Aves: Furnariidae) Б/ Major Shifts in Nest Architecture and Adaptive Radiatio
JOURNAL OF AVIAN BIOLOGY 37: 260Á/272, 2006 Evolution of the ovenbird-woodcreeper assemblage (Aves: Furnariidae) / major shifts in nest architecture and adaptive radiation Á Martin Irestedt, Jon Fjeldsa˚ and Per G. P. Ericson Irestedt, M., Fjeldsa˚, J. and Ericson, P. G. P. 2006. Evolution of the ovenbird- woodcreeper assemblage (Aves: Furnariidae) Á/ major shifts in nest architecture and adaptive radiation. Á/ J. Avian Biol. 37: 260Á/272 The Neotropical ovenbirds (Furnariidae) form an extraordinary morphologically and ecologically diverse passerine radiation, which includes many examples of species that are superficially similar to other passerine birds as a resulting from their adaptations to similar lifestyles. The ovenbirds further exhibits a truly remarkable variation in nest types, arguably approaching that found in the entire passerine clade. Herein we present a genus-level phylogeny of ovenbirds based on both mitochondrial and nuclear DNA including a more complete taxon sampling than in previous molecular studies of the group. The phylogenetic results are in good agreement with earlier molecular studies of ovenbirds, and supports the suggestion that Geositta and Sclerurus form the sister clade to both core-ovenbirds and woodcreepers. Within the core-ovenbirds several relationships that are incongruent with traditional classifications are suggested. Among other things, the philydorine ovenbirds are found to be non-monophyletic. The mapping of principal nesting strategies onto the molecular phylogeny suggests cavity nesting to be plesiomorphic within the ovenbirdÁ/woodcreeper radiation. It is also suggested that the shift from cavity nesting to building vegetative nests is likely to have happened at least three times during the evolution of the group. -

Threatened Birds of the Americas
GREY-HEADED ANTBIRD Myrmeciza griseiceps E2 This rare formicariid is confined to patches of bamboo and dense undergrowth in semi-deciduous moist forest and cloud-forest in the Pacific slope foothills of the Andes in south-west Ecuador and north-west Peru, where it is threatened by habitat destruction. DISTRIBUTION The Grey-headed Antbird (see Remarks 1) is confined to the Pacific slope of the Andes in El Oro and Loja provinces, south-west Ecuador, and Tumbes and Piura departments, north-west Peru, where it is found at elevations ranging from 600 to 2,900 m. The bird is known from few specimens, and is generally distributed in five areas, where localities (coordinates from Paynter and Traylor 1977 and Stephens and Traylor 1983) are as follows: Ecuador (El Oro) La Chonta, 610 m (Chapman 1926, Zimmer 1932), at 3°35’S 79°53’W (see Remarks 2); San Pablo, 1,200 m, at 3°41’S 79°33’W, near Zaruma, where a bird (possibly this species) was heard calling in 1991 (Williams and Tobias 1991); (Loja) above Vicentino, 1,250-1,450 m, at c.3°56’S 79°55’W (five males singing in February 1991: Best 1992); Alamor, 1,400 m (Chapman 1926) at 4°02’S 80°02’W; Celica, 2,100 m (Zimmer 1932) at 4°07’S 79°59’W; 8 km west of Celica, 1,900 m (one specimen collected and others seen in August 1989: R. S. Ridgely in litt. 1989); near San José de Pozul along the road to Pindal, 1,600 m, at 4°07’S 80°03’W (sightings in April–May 1989: P. -

Phylogenetic Analysis of the Nest Architecture of Neotropical Ovenbirds (Furnariidae)
The Auk 116(4):891-911, 1999 PHYLOGENETIC ANALYSIS OF THE NEST ARCHITECTURE OF NEOTROPICAL OVENBIRDS (FURNARIIDAE) KRZYSZTOF ZYSKOWSKI • AND RICHARD O. PRUM NaturalHistory Museum and Department of Ecologyand Evolutionary Biology, University of Kansas,Lawrence, Kansas66045, USA ABSTRACT.--Wereviewed the tremendousarchitectural diversity of ovenbird(Furnari- idae) nestsbased on literature,museum collections, and new field observations.With few exceptions,furnariids exhibited low intraspecificvariation for the nestcharacters hypothe- sized,with the majorityof variationbeing hierarchicallydistributed among taxa. We hy- pothesizednest homologies for 168species in 41 genera(ca. 70% of all speciesand genera) and codedthem as 24 derivedcharacters. Forty-eight most-parsimonious trees (41 steps,CI = 0.98, RC = 0.97) resultedfrom a parsimonyanalysis of the equallyweighted characters using PAUP,with the Dendrocolaptidaeand Formicarioideaas successiveoutgroups. The strict-consensustopology based on thesetrees contained 15 cladesrepresenting both tra- ditionaltaxa and novelphylogenetic groupings. Comparisons with the outgroupsdemon- stratethat cavitynesting is plesiomorphicto the furnariids.In the two lineageswhere the primitivecavity nest has been lost, novel nest structures have evolved to enclosethe nest contents:the clayoven of Furnariusand the domedvegetative nest of the synallaxineclade. Althoughour phylogenetichypothesis should be consideredas a heuristicprediction to be testedsubsequently by additionalcharacter evidence, this first cladisticanalysis -
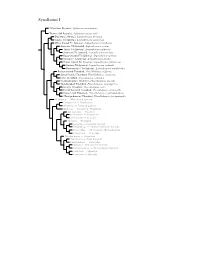
Synallaxini Species Tree
Synallaxini I ?Masafuera Rayadito, Aphrastura masafuerae Thorn-tailed Rayadito, Aphrastura spinicauda Des Murs’s Wiretail, Leptasthenura desmurii Tawny Tit-Spinetail, Leptasthenura yanacensis White-browed Tit-Spinetail, Leptasthenura xenothorax Araucaria Tit-Spinetail, Leptasthenura setaria Tufted Tit-Spinetail, Leptasthenura platensis Striolated Tit-Spinetail, Leptasthenura striolata Rusty-crowned Tit-Spinetail, Leptasthenura pileata Streaked Tit-Spinetail, Leptasthenura striata Brown-capped Tit-Spinetail, Leptasthenura fuliginiceps Andean Tit-Spinetail, Leptasthenura andicola Plain-mantled Tit-Spinetail, Leptasthenura aegithaloides Rufous-fronted Thornbird, Phacellodomus rufifrons Streak-fronted Thornbird, Phacellodomus striaticeps Little Thornbird, Phacellodomus sibilatrix Chestnut-backed Thornbird, Phacellodomus dorsalis Spot-breasted Thornbird, Phacellodomus maculipectus Greater Thornbird, Phacellodomus ruber Freckle-breasted Thornbird, Phacellodomus striaticollis Orange-eyed Thornbird, Phacellodomus erythrophthalmus ?Orange-breasted Thornbird, Phacellodomus ferrugineigula Hellmayrea — White-tailed Spinetail Coryphistera — Brushrunner Anumbius — Firewood-gatherer Asthenes — Canasteros, Thistletails Acrobatornis — Graveteiro Metopothrix — Plushcrown Xenerpestes — Graytails Siptornis — Prickletail Roraimia — Roraiman Barbtail Thripophaga — Speckled Spinetail, Softtails Limnoctites — ST Spinetail, SB Reedhaunter Cranioleuca — Spinetails Pseudasthenes — Canasteros Spartonoica — Wren-Spinetail Pseudoseisura — Cacholotes Mazaria – White-bellied -

Geographic Variation, Zoogeography, and Possible Rapid Evolution in Some Cranioleuca Spinetails (Furnariidae) of the Andes
THE WILSON BULLETIN A QUARTERLY MAGAZINE OF ORNITHOLOGY Published by the Wilson Ornithological Society VOL. 96, No. 4 DECEMBER 1984 PAGES 5 15-775 Wilson Bull., 96(4), 1984, pp. 5 15-523 GEOGRAPHIC VARIATION, ZOOGEOGRAPHY, AND POSSIBLE RAPID EVOLUTION IN SOME CRANIOLEUCA SPINETAILS (FURNARIIDAE) OF THE ANDES J. V. REMSEN, JR. The humid slopes of the Andes Mountains of South America provide one of the worlds’ greatest natural laboratories for the study of evolution and zoogeography. Although the “population structure,” nature of geo- graphic variation, and location of zoogeographic boundaries have been described for numerous taxa of the puna and paramo zones of the Andes (Vuilleumier 1980) relatively little has been published concerning birds of the humid, forested eastern slopes. In this paper, I analyze the geo- graphic variation and distribution of the Cranioleuca albiceps superspe- ties (Fumariidae), here considered to consist of Light-crowned Spinetail (C. a/biceps) (with two subspecies), Marcapata Spinetail (C. marcapatae), and a previously undescribed form, here considered a subspecies of C. marcapatae, to be called: Cranioleuca marcapatae weskeisubsp. nov. HOLOTYPE.-American Museum of Natural History No. 820557; male from Cordillera Vilcabamba, elev. 3250 m, Dpto. Cuzco, Peru (12”36S,’ 733O’ W),’ 22 July 1968; John S. Weske, original number 1825. DIAGNOSIS.-Ventrally very similar to Crunioleuca m. marcupatae, but malar region more strongly washed buff; dorsally, virtually identical to white-crowned individuals of Cranioleuca a.