Integrated Deep Learned Transcriptomic and Structure-Based Predictor of Clinical Trials Outcomes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Applying Screening Techniques to Two Orphan Gpcrs
Universidade de Lisboa Faculdade de Farmácia Deorphanization of receptors: Applying screening techniques to two orphan GPCRs Ana Catarina Rufas da Silva Santos Mestrado Integrado em Ciências Farmacêuticas 2019 Universidade de Lisboa Faculdade de Farmácia Deorphanization of receptors: Applying screening techniques to two orphan GPCRs Ana Catarina Rufas da Silva Santos Monografia de Mestrado Integrado em Ciências Farmacêuticas apresentada à Universidade de Lisboa através da Faculdade de Farmácia Orientadora: Ghazl Al Hamwi, PhD Student Co-Orientadora: Professora Doutora Elsa Maria Ribeiro dos Santos Anes, Professora Associada com Agregação em Microbiologia 2019 Abstract G-Protein Coupled Receptors represent one of the largest families of cellular receptors discovered and one of the main sources of attractive drug targets. In contrast, it also has a large number of understudied or orphan receptors. Pharmacological assays such as β-Arrestin recruitment assays, are one of the possible approaches for deorphanization of receptors. In this work, I applied the assay system previously mentioned to screen compounds in two orphan receptors, GRP37 and MRGPRX3. GPR37 has been primarily associated with a form of early onset Parkinsonism due to its’ expression patterns, and physiological role as substrate to ubiquitin E3, parkin. Although extensive literature regarding this receptor is available, the identification of a universally recognized ligand has not yet been possible. Two compounds were proposed as ligands, but both were met with controversy. These receptor association with Autosomal Recessive Juvenile Parkinson positions it as a very attractive drug target, and as such its’ deorphanization is a prime objective for investigators in this area. Regarding MRGPRX3 information is much scarcer. -

Preladenant | Medchemexpress
Inhibitors Product Data Sheet Preladenant • Agonists Cat. No.: HY-10889 CAS No.: 377727-87-2 Molecular Formula: C₂₅H₂₉N₉O₃ • Molecular Weight: 503.56 Screening Libraries Target: Adenosine Receptor Pathway: GPCR/G Protein Storage: Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month SOLVENT & SOLUBILITY In Vitro DMSO : 5 mg/mL (9.93 mM; Need ultrasonic) Mass Solvent 1 mg 5 mg 10 mg Concentration Preparing 1 mM 1.9859 mL 9.9293 mL 19.8586 mL Stock Solutions 5 mM 0.3972 mL 1.9859 mL 3.9717 mL 10 mM --- --- --- Please refer to the solubility information to select the appropriate solvent. In Vivo 1. Add each solvent one by one: 10% DMSO >> 40% PEG300 >> 5% Tween-80 >> 45% saline Solubility: 0.5 mg/mL (0.99 mM); Suspended solution; Need ultrasonic 2. Add each solvent one by one: 10% DMSO >> 90% (20% SBE-β-CD in saline) Solubility: 0.5 mg/mL (0.99 mM); Suspended solution; Need ultrasonic 3. Add each solvent one by one: 10% DMSO >> 90% corn oil Solubility: ≥ 0.5 mg/mL (0.99 mM); Clear solution BIOLOGICAL ACTIVITY Description Preladenant is a potent and competitive antagonist of the human adenosine A2A receptor with a Ki of 1.1 nM and has over 1000-fold selectivity over other adenosine receptors. [1] IC₅₀ & Target Ki: 1.1 nM (Adenosine A2A receptor) In Vitro Preladenant also completely antagonizes cAMP in cells expressing the recombinant human A2A receptor. Preladenant is determined to has KB values of 1.3 nM at the A2A receptor; the value is in good agreement with the Ki value determined in Page 1 of 3 www.MedChemExpress.com the radioligand binding assay. -

1.Intro Corregida
Heterómeros de receptores de dopamina como dianas terapéuticas en la enfermedad de Parkinson y en discinesias inducidas por el tratamiento con L-DOPA Daniel Farré Pérez ADVERTIMENT. La consulta d’aquesta tesi queda condicionada a l’acceptació de les següents condicions d'ús: La difusió d’aquesta tesi per mitjà del servei TDX (www.tdx.cat) i a través del Dipòsit Digital de la UB (diposit.ub.edu) ha estat autoritzada pels titulars dels drets de propietat intel·lectual únicament per a usos privats emmarcats en activitats d’investigació i docència. No s’autoritza la seva reproducció amb finalitats de lucre ni la seva difusió i posada a disposició des d’un lloc aliè al servei TDX ni al Dipòsit Digital de la UB. No s’autoritza la presentació del seu contingut en una finestra o marc aliè a TDX o al Dipòsit Digital de la UB (framing). Aquesta reserva de drets afecta tant al resum de presentació de la tesi com als seus continguts. En la utilització o cita de parts de la tesi és obligat indicar el nom de la persona autora. ADVERTENCIA. La consulta de esta tesis queda condicionada a la aceptación de las siguientes condiciones de uso: La difusión de esta tesis por medio del servicio TDR (www.tdx.cat) y a través del Repositorio Digital de la UB (diposit.ub.edu) ha sido autorizada por los titulares de los derechos de propiedad intelectual únicamente para usos privados enmarcados en actividades de investigación y docencia. No se autoriza su reproducción con finalidades de lucro ni su difusión y puesta a disposición desde un sitio ajeno al servicio TDR o al Repositorio Digital de la UB. -

The Two Tondition to Martin
THETWO TONDITIONUS 20170306038A1 TO MARTIN ( 19) United States (12 ) Patent Application Publication (10 ) Pub. No. : US 2017/ 0306038 A1 BROGDON et al. (43 ) Pub . Date : Oct. 26 , 2017 ( 54 ) COMPOSITIONS AND METHODS OF USE Publication Classification FOR AUGMENTED IMMUNE RESPONSE (51 ) Int. Cl. AND CANCER THERAPY CO7K 16 / 28 ( 2006 .01 ) (71 ) Applicants : Jennifer BROGDON , Sudbury , MA CO7K 16 / 28 ( 2006 . 01) (US ) ; Daniela CIPOLLETTA , A61K 45 / 06 (2006 . 01 ) Arlington , MA (US ) ; Glenn A61K 39 /00 ( 2006 .01 ) DRANOFF , Lexington , MA ( US ) ; A61K 39 /00 (2006 .01 ) Deborah A . KNEE . Del Mar. CA (US ): (52 ) U . S . Cl. CPC . .. .. CO7K 16 / 2878 ( 2013. 01 ) ; C07K 16 /2818 Fei WANG , San Diego , CA (US ) ( 2013 .01 ) ; A61K 45 / 06 ( 2013 .01 ) ; CO7K ( 72 ) Inventors : Jennifer BROGDON , Sudbury , MA 2317 / 74 ( 2013 . 01 ) ; C07K 2317 / 732 ( 2013 .01 ) ; (US ) ; Daniela CIPOLLETTA , CO7K 2317 / 567 ( 2013 . 01 ) ; COZK 2317 / 56 Arlington , MA (US ) ; Glenn ( 2013 .01 ) ; CO7K 2317 /51 ( 2013 .01 ) ; CO7K DRANOFF , Lexington , MA (US ) ; 2317 / 24 (2013 .01 ) ; A61K 2039 /507 (2013 . 01 ) ; Deborah A . KNEE , Del Mar, CA (US ) ; A61K 2039 /505 ( 2013 . 01 ) ; COZK 2317 / 75 ( 2013 . 01 ) ; CO7K 2317/ 92 ( 2013 .01 ) ; CO7K Fei WANG , San Diego , CA (US ) 2317/ 515 (2013 . 01) ( 21 ) Appl. No. : 15 /517 ,872 (57 ) ABSTRACT (22 ) PCT Filed : Oct . 8 , 2015 The present invention provides antibody compositions , (86 ) PCT No. : PCT/ US2015 / 054770 including , e . g ., antibodies, engineered antibodies and anti body fragments that bind to a tumor necrosis factor receptor $ 371 ( c ) ( 1 ), superfamily member ( i. e . , 18 ) , and compositions comprising ( 2 ) Date : Apr. -

(12) United States Patent (10) Patent No.: US 9.402,830 B2 Cialella Et Al
USOO940283OB2 (12) United States Patent (10) Patent No.: US 9.402,830 B2 Cialella et al. (45) Date of Patent: * Aug. 2, 2016 (54) METHODS OF TREATING DYSKINESIA AND FOREIGN PATENT DOCUMENTS RELATED DSORDERS WO 93.18767 A1 9, 1993 (71) Applicant: Melior Discovery, Inc., Exton, PA (US) OTHER PUBLICATIONS (72) Inventors: John Ciallella, Exton, PA (US); John Afanasev et al., Effects of amphetamine and Sydnocarbon dopamine Gruner, Exton, PA (US); Andrew G. release and free radical generation in rat striatum, Pharmacol Reaume, Exton, PA (US); Michael S. Biochem Behav, 2001, 69(3-4):653-8. Saporito, Exton, PA (US) Anderzhanova et al., Effect of d-amphetamine and Sydnocarb on the extracellular level of dopamine, 3,4-dihydroxyphenylacetic acid, and (73) Assignee: Melior Discovery, Inc., Exton, PA (US) hydroxyl radicals generation in rat striatum, Ann NY AcadSci, 2000, 914:137-45. Anderzhanova et al., Effects of Sydnocarb and D-amphetamine on the (*) Notice: Subject to any disclaimer, the term of this extracellular levels of amino acids in the rat caudate-putamen, Eur J patent is extended or adjusted under 35 Pharmacol, 2001, 428(1):87-95. U.S.C. 154(b) by 0 days. Bashkatova et al., Neuroshemical changes and neurotoxic effects of This patent is Subject to a terminal dis an acute treatment with Sydnocarb, a novel psychostimulant: com claimer. parison with D-amphetamine, Ann NY AcadSci, 2002,965: 180-192. Cody, Precursor medications as a source of methamphetamine and/or amphetamine positive drug testing results, J Occup Environ Med, (21) Appl. No.: 14/702,242 2002, 44(5):435-50. -

Chemical Properties Biological Description
Data Sheet (Cat.No.T4290) Preladenant Chemical Properties CAS No.: 377727-87-2 Formula: C25H29N9O3 Molecular Weight: 503.57 Appearance: N/A Storage: 0-4℃ for short term (days to weeks), or -20℃ for long term (months). Biological Description Description Preladenant is an orally bioavailable antagonist of the adenosine A2A receptor (Ki: 1.1 nM) and has >1000-fold selectivity over all other adenosine receptors. Targets(IC50) A2A: None In vitro In cells expressing the recombinant human A2A receptor, Preladenant completely antagonizes cAMP. The KB values of Preladenant is 1.3 nM at the A2A receptor. A similar functional assay with A2B receptor-expressing cells is used to demonstrate selectivity over A2B receptors. In this assay, the KB value for Preladenant is 1.2 μM and this selectivity for the A2A receptor is 923-fold over the A2B receptor. In vivo Preladenant (1 mg/kg) inhibits L-Dopa-induced behavioral sensitization. In the mouse tail suspension test and the mouse and rat forced swim test, Preladenant exhibits antidepressant-like profiles. Preladenant dose- dependently reduces the parkinsonian scores at doses of 1 mg/kg (min score: 9.0) and 3 mg/kg (min score: 6.5). A subthreshold dose of Preladenant reduces minimum and mean parkinsonian scores in animals treated with 3 mg kg of L-Dopa to 5.25 and 6.88 respectively. A Wilcoxin test is used to compare individual treatments against the vehicle. Preladenant (3 mg/kg), L-Dopa (3, 6, and 12 mg/kg), and the combination of Preladenant and L- Dopa (1 or 3 mg/kg+3 mg/kg) are all significantly improved on the minimum parkinsonian score. -

Advances in Non-Dopaminergic Treatments for Parkinson's Disease
REVIEW ARTICLE published: 22 May 2014 doi: 10.3389/fnins.2014.00113 Advances in non-dopaminergic treatments for Parkinson’s disease Sandy Stayte 1,2 and Bryce Vissel 1,2* 1 Neuroscience Department, Neurodegenerative Disorders Laboratory, Garvan Institute of Medical Research, Sydney, NSW, Australia 2 Faculty of Medicine, University of New South Wales, Sydney, NSW, Australia Edited by: Since the 1960’s treatments for Parkinson’s disease (PD) have traditionally been directed Eero Vasar, University of Tartu, to restore or replace dopamine, with L-Dopa being the gold standard. However, chronic Estonia L-Dopa use is associated with debilitating dyskinesias, limiting its effectiveness. This has Reviewed by: resulted in extensive efforts to develop new therapies that work in ways other than Andrew Harkin, Trinity College Dublin, Ireland restoring or replacing dopamine. Here we describe newly emerging non-dopaminergic Sulev Kõks, University of Tartu, therapeutic strategies for PD, including drugs targeting adenosine, glutamate, adrenergic, Estonia and serotonin receptors, as well as GLP-1 agonists, calcium channel blockers, iron Pille Taba, Universoty of Tartu, chelators, anti-inflammatories, neurotrophic factors, and gene therapies. We provide a Estonia Pekka T. Männistö, University of detailed account of their success in animal models and their translation to human clinical Helsinki, Finland trials. We then consider how advances in understanding the mechanisms of PD, genetics, *Correspondence: the possibility that PD may consist of multiple disease states, understanding of the Bryce Vissel, Neuroscience etiology of PD in non-dopaminergic regions as well as advances in clinical trial design Department, Neurodegenerative will be essential for ongoing advances. We conclude that despite the challenges ahead, Disorders Laboratory, Garvan Institute of Medical Research, patients have much cause for optimism that novel therapeutics that offer better disease 384 Victoria Street, Darlinghurst, management and/or which slow disease progression are inevitable. -

5'-Nucleotidases, Nucleosides and Their Distribution in the Brain: Patho- Logical and Therapeutic Implications
Send Orders for Reprints to [email protected] Current Medicinal Chemistry, 2013, 20, 4217-4240 4217 5'-Nucleotidases, Nucleosides and their Distribution in the Brain: Patho- logical and Therapeutic Implications Zsolt Kovács1,*, Árpád Dobolyi2, Katalin A. Kékesi3,4 and Gábor Juhász3 1Department of Zoology, University of West Hungary, Savaria Campus, Szombathely, Hungary; 2Laboratory of Neuro- morphology, Department of Anatomy, Histology and Embryology, Semmelweis University and the Hungarian Academy of Sciences, Budapest, Hungary; 3Laboratory of Proteomics, Institute of Biology, Eötvös Loránd University, Budapest, Hungary; 4 Department of Physiology and Neurobiology, Eötvös Loránd University, Budapest, Hungary Abstract: Elements of the nucleoside system (nucleoside levels, 5’-nucleotidases (5’NTs) and other nucleoside metabolic enzymes, nucleoside transporters and nucleoside receptors) are unevenly distributed in the brain, suggesting that nucleo- sides have region-specific functions in the human brain. Indeed, adenosine (Ado) and non-Ado nucleosides, such as guanosine (Guo), inosine (Ino) and uridine (Urd), modulate both physiological and pathophysiological processes in the brain, such as sleep, pain, memory, depression, schizophrenia, epilepsy, Huntington’s disease, Alzheimer’s disease and Parkinson’s disease. Interactions have been demonstrated in the nucleoside system between nucleoside levels and the ac- tivities of nucleoside metabolic enzymes, nucleoside transporters and Ado receptors in the human brain. Alterations in the nucleoside system may induce pathological changes, resulting in central nervous system (CNS) diseases. Moreover, sev- eral CNS diseases such as epilepsy may be treated by modulation of the nucleoside system, which is best achieved by modulating 5’NTs, as 5’NTs exhibit numerous functions in the CNS, including intracellular and extracellular formation of nucleosides, termination of nucleoside triphosphate signaling, cell adhesion, synaptogenesis and cell proliferation. -

Biomolecules
biomolecules Review Receptor Ligands as Helping Hands to L-DOPA in the Treatment of Parkinson’s Disease Fabio Del Bello , Mario Giannella, Gianfabio Giorgioni * , Alessandro Piergentili and Wilma Quaglia Scuola di Scienze del Farmaco e dei Prodotti della Salute, Università di Camerino, Via S. Agostino 1, 62032 Camerino, Italy; [email protected] (F.D.B.); [email protected] (M.G.); [email protected] (A.P.); [email protected] (W.Q.) * Correspondence: [email protected]; Tel.: +39-0737402368 Received: 18 March 2019; Accepted: 6 April 2019; Published: 9 April 2019 Abstract: Levodopa (LD) is the most effective drug in the treatment of Parkinson’s disease (PD). However, although it represents the “gold standard” of PD therapy, LD can cause side effects, including gastrointestinal and cardiovascular symptoms as well as transient elevated liver enzyme levels. Moreover, LD therapy leads to LD-induced dyskinesia (LID), a disabling motor complication that represents a major challenge for the clinical neurologist. Due to the many limitations associated with LD therapeutic use, other dopaminergic and non-dopaminergic drugs are being developed to optimize the treatment response. This review focuses on recent investigations about non-dopaminergic central nervous system (CNS) receptor ligands that have been identified to have therapeutic potential for the treatment of motor and non-motor symptoms of PD. In a different way, such agents may contribute to extending LD response and/or ameliorate LD-induced side effects. Keywords: Parkinson’s disease; levodopa therapy; levodopa-induced side effects; dopaminergic drugs; non-dopaminergic receptor ligands 1. Introduction Parkinson’s disease (PD), also known as idiopathic paralysis agitans, is one of the most frequent chronic neurodegenerative diseases worldwide. -

Unprecedented Therapeutic Potential with a Combination of A2A/NR2B Receptor Antagonists As Observed in the 6-OHDA Lesioned Rat Model of Parkinson’S Disease
RESEARCH ARTICLE Unprecedented Therapeutic Potential with a Combination of A2A/NR2B Receptor Antagonists as Observed in the 6-OHDA Lesioned Rat Model of Parkinson’s Disease Anne Michel1*, Patrick Downey1, Jean-Marie Nicolas2, Dieter Scheller1 1. Neurosciences TA Biology, UCB BioPharma SPRL, Braine l’Alleud, Belgium, 2. Non-Clinical Development, UCB BioPharma SPRL, Braine l’Alleud, Belgium *[email protected] OPEN ACCESS Citation: Michel A, Downey P, Nicolas J-M, Scheller D (2014) Unprecedented Therapeutic Potential with a Combination of A2A/NR2B Receptor Antagonists as Observed in the 6-OHDA Abstract Lesioned Rat Model of Parkinson’s Disease. PLoS ONE 9(12): e114086. doi:10.1371/journal.pone. In Parkinson’s disease, the long-term use of dopamine replacing agents is 0114086 associated with the development of motor complications; therefore, there is a need Editor: Joohyung Lee, Prince Henry’s Institute, Australia for non-dopaminergic drugs. This study evaluated the potential therapeutic impact Received: July 17, 2014 of six different NR2B and A2A receptor antagonists given either alone or in Accepted: November 4, 2014 combination in unilateral 6-OHDA-lesioned rats without (monotherapy) or with (add- on therapy) the co-administration of L-Dopa: Sch-58261+ Merck 22; Sch- Published: December 16, 2014 58261+Co-101244; Preladenant + Merck 22; Preladenant + Radiprodil; Tozadenant Copyright: ß 2014 Michel et al. This is an open- access article distributed under the terms of the + Radiprodil; Istradefylline + Co-101244. Animals given monotherapy were Creative Commons Attribution License, which permits unrestricted use, distribution, and repro- assessed on distance traveled and rearing, whereas those given add-on therapy duction in any medium, provided the original author were assessed on contralateral rotations. -

Adenosinrezeptoren Auf Humanen T-Lymphozyten: Modulation Durch Subtyp-Selektive Rezeptor- Agonisten Und -Antagonisten
Adenosinrezeptoren auf humanen T-Lymphozyten: Modulation durch Subtyp-selektive Rezeptor- Agonisten und -Antagonisten Dissertation zur Erlangung des Doktorgrades (Dr. rer. nat.) der Mathematisch-Naturwissenschaftlichen Fakultät der Rheinischen Friedrich-Wilhelms-Universität Bonn vorgelegt von Svenja K. Lacher aus Villingen-Schwenningen Bonn 2009 Angefertigt mit Genehmigung der Mathematisch-Naturwissenschaftlichen Fakultät der Rheinischen Friedrich-Wilhelms-Universität Bonn 1. Referent: Prof. Dr. Christa E. Müller 2. Referent: Prof. Dr. Gerd Bendas 3. Referent: Prof. Dr. Gabriele König 4. Referent: Prof. Dr. Hanns Häberlein Tag der Promotion: 20. März 2009 Diese Dissertation ist auf dem Hochschulserver der ULB Bonn http://hss.ulb.uni- bonn.de/diss-online elektronisch publiziert. Die vorliegende Arbeit wurde in der Zeit von April 2005 bis Dezember 2008 am Pharma- zeutischen Institut der Rheinischen Friedrich-Wilhelms-Universität Bonn unter der Leitung von Frau Prof. Dr. Christa E. Müller durchgeführt. Mein besonderer Dank gilt Frau Prof. Dr. Christa E. Müller, die mir durch ihre Unterstüt- zung die erfolgreiche Absolvierung des Qualifizierungsjahres für Fachhochschulabsolven- ten ermöglicht hat, und mir dadurch die Möglichkeit bot, diese Arbeit mit der interessanten Themenstellung anzufertigen. Ich danke ihr für die stets freundliche Betreuung, die Dis- kussionsbereitschaft und die zahlreichen Anregungen, die maßgeblich zum Gelingen dieser Arbeit beigetragen haben. Herrn Prof. Dr. Gerd Bendas danke ich für die freundliche Übernahme des Koreferates; Frau Prof. Dr. Gabriele König und Herrn Prof. Dr. Hanns Häberlein danke ich für die Mitwirkung in meiner Promotionskommision. Ich danke der deutschen Forschungsgemeinschaft für die finanzielle Unterstützung in Form eines Stipendiums für die Heranführung an die Doktorarbeit und eines Promotionsstipen- diums im Rahmen des Graduiertenkollegs 677 „Struktur und molekulare Interaktion als Basis der Arzneimittelwirkung“. -
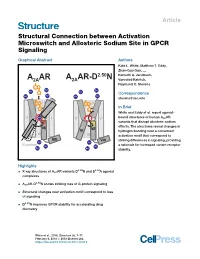
Structural Connection Between Activation Microswitch and Allosteric Sodium Site in GPCR Signaling
Article Structural Connection between Activation Microswitch and Allosteric Sodium Site in GPCR Signaling Graphical Abstract Authors Kate L. White, Matthew T. Eddy, Zhan-Guo Gao, ..., A AR A AR-D2.50N Kenneth A. Jacobson, 2A 2A Vsevolod Katritch, Raymond C. Stevens Na+ Na+ Na+ Na+ Correspondence Na+ Na+ Na+ Na+ [email protected] II II In Brief White and Eddy et al. report agonist- V Na+ I V I bound structures of human A2AAR variants that disrupt allosteric sodium VI VII VII effects. The structures reveal changes in VI hydrogen bonding near a conserved III III activation motif that correspond to VIII VIII striking differences in signaling, providing Na+ Na+ G-protein Na+ Na+ a rationale for increased variant receptor Na+ Na+ stability. Highlights 2.50 3.39 d X-ray structures of A2AAR variants D N and S A agonist complexes 2.50 d A2AAR-D N shows striking loss of G-protein signaling d Structural changes near activation motif correspond to loss of signaling d D2.50N improves GPCR stability for accelerating drug discovery White et al., 2018, Structure 26, 1–11 February 6, 2018 ª 2018 Elsevier Ltd. https://doi.org/10.1016/j.str.2017.12.013 Please cite this article in press as: White et al., Structural Connection between Activation Microswitch and Allosteric Sodium Site in GPCR Signaling, Structure (2018), https://doi.org/10.1016/j.str.2017.12.013 Structure Article Structural Connection between Activation Microswitch and Allosteric Sodium Site in GPCR Signaling Kate L. White,1,3 Matthew T. Eddy,1,3 Zhan-Guo Gao,2 Gye Won Han,1 Tiffany Lian,1 Alexander Deary,1 Nilkanth Patel,1 Kenneth A.