Split Spinal Cord Malformations in Children
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Split Notochord Syndrome
0021-7557/04/80-01/77 Jornal de Pediatria Copyright © 2004 by Sociedade Brasileira de Pediatria RELATO DE CASO Síndrome do notocórdio fendido, variante rara do cisto neuroentérico A rare variant of neuroenteric cyst: split notochord syndrome Lisieux E. Jesus1, Cristiano G. França2 Resumo Abstract Objetivo: Estudo de um caso de síndrome do notocórdio fendido, Objective: We present a case of split notochord syndrome, an forma extremamente rara de disrafismo medular. A literatura pertinen- extremely rare form of spinal dysraphism. te, pesquisada através das bases de dados MEDLINE e LILACS, é Description: We treated a 2 month-old boy presenting with an analisada e sumarizada. extensive lumbosacral deformity, hydrocephalus and apparent enteric Descrição: Foi atendido lactente masculino de 2 meses de idade segments in the dorsal midline, accompanied by an enteric fistula apresentando extensa deformidade de coluna lombo-sacra, hidrocefa- and imperforated anus. The malformation was diagnosed as split lia e exteriorização de alças intestinais pela linha média dorsal, notochord syndrome. The baby died as a result of sepsis before acompanhada de fístula entérica e imperfuração anal. A malformação surgical treatment could be attempted. foi diagnosticada como síndrome do notocórdio fendido. A criança Comments: Split notochord syndrome is the rarest form of evoluiu para óbito secundário a sepse antes de ser feito qualquer neuroenteric cyst described until this moment (<25 cases in the tratamento cirúrgico. literature). It is frequently associated with anorectal malformation, Comentários: A síndrome do notocórdio fendido é a forma mais intestinal fistulae and hydrocephalus. Prognosis is not necessarily rara de cisto neuroentérico já descrita (<25 casos descritos em poor and survival is possible if digestive malformations, hydrocephalus literatura) e está associada freqüentemente a fístulas digestivas, and the dysraphism itself are treated simultaneously. -

Intramedullary Cystic Lesions Ofthe Conus Medullaris
J Neurol Neurosurg Psychiatry: first published as 10.1136/jnnp.31.2.106 on 1 April 1968. Downloaded from J. Neurol. Neurosurg. Psychiat., 1968, 31, 106-109 Intramedullary cystic lesions of the conus medullaris SAMI I. NASSAR, JAMES W. CORRELL, AND EDGAR M. HOUSEPIAN From the Department of Neurosurgery, College ofPhysicians and Surgeons, Columbia University, and the Neurological Institute of the Columbia-Presbyterian Medical Center, New York, U.S.A. Intramedullary cystic lesions of the conus medullaris of the aetiology, these cysts may simulate the clinical are rare. Although an extensive literature describes picture of syringomyelia. syringomyelia as being a frequent basis for cystic The cases of cysts of the conus medullaris re- cervico-thoracic lesions it is apparent that this ported here simulated the clinical picture of does not occur frequently in the lumbosacral region syringomyelia, tumour, or lumbar disc disease. (Kirgis and Echols, 1949; Netsky, 1953; Rand and The radiographic findings in each case were inter- Rand, 1960; Love and Olafson, 1966). Poser (1956), preted as indicating the presence ofan intramedullary in a review of 234 cases of syringomyelia, found tumour. The correct diagnosis was made in each that the cavity extended into the lumbosacral region case only at operation. in only 12-6% and in only five cases were the Protected by copyright. cavities restricted to the lumbosacral segments. Some authors (Thevenard, 1942; Andre, 1951) CASE REPORTS question the occurrence of syringomyelia in the lower spinal cord. Nevertheless a high incidence CASE 1 (F.T., NO. 179 16 92) A 22-year-old negro male of constitutional defects has been noted among was admitted complaining of weakness and pain in the syringomyelia patients and members of their legs for three years. -

The Spinal Cord Is a Nerve Column That Passes Downward from Brain Into the Vertebral Canal
The spinal cord is a nerve column that passes downward from brain into the vertebral canal. Recall that it is part of the CNS. Spinal nerves extend to/from the spinal cord and are part of the PNS. Length = about 17 inches Start = foramen magnum End = tapers to point (conus medullaris) st nd and terminates 1 –2 lumbar (L1-L2) vertebra Contains 31 segments à gives rise to 31 pairs of spinal nerves Note cervical and lumbar enlargements. cauda equina (“horse’s tail”) –collection of spinal nerves at inferior end of vertebral column (nerves coming off end of spinal cord) Meninges- cushion and protected by same 3 layers as brain. Extend past end of cord into vertebral canal à spinal tap because no cord A cross-section of the spinal cord resembles a butterfly with its wings outspread (gray matter) surrounded by white matter. GRAY MATTER or “butterfly” = bundles of cell bodies Posterior (dorsal) horns=association or interneurons (incoming somatosensory information) Lateral horns=autonomic neurons Anterior (ventral) horns=cell bodies of motor neurons Central canal-found within gray matter and filled with CSF White Matter: 3 Regions: Posterior (dorsal) white column or funiculi – contains only ASCENDING tracts à sensory only Lateral white column or funiculi – both ascending and descending tracts à sensory and motor Anterior (ventral) white column or funiculi – both ascending and descending tracts à sensory and motor All nerve tracts made of mylinated axons with same destination and function Associated Structures: Dorsal Roots = made -

Spinal Cord Organization
Lecture 4 Spinal Cord Organization The spinal cord . Afferent tract • connects with spinal nerves, through afferent BRAIN neuron & efferent axons in spinal roots; reflex receptor interneuron • communicates with the brain, by means of cell ascending and descending pathways that body form tracts in spinal white matter; and white matter muscle • gives rise to spinal reflexes, pre-determined gray matter Efferent neuron by interneuronal circuits. Spinal Cord Section Gross anatomy of the spinal cord: The spinal cord is a cylinder of CNS. The spinal cord exhibits subtle cervical and lumbar (lumbosacral) enlargements produced by extra neurons in segments that innervate limbs. The region of spinal cord caudal to the lumbar enlargement is conus medullaris. Caudal to this, a terminal filament of (nonfunctional) glial tissue extends into the tail. terminal filament lumbar enlargement conus medullaris cervical enlargement A spinal cord segment = a portion of spinal cord that spinal ganglion gives rise to a pair (right & left) of spinal nerves. Each spinal dorsal nerve is attached to the spinal cord by means of dorsal and spinal ventral roots composed of rootlets. Spinal segments, spinal root (rootlets) nerve roots, and spinal nerves are all identified numerically by th region, e.g., 6 cervical (C6) spinal segment. ventral Sacral and caudal spinal roots (surrounding the conus root medullaris and terminal filament and streaming caudally to (rootlets) reach corresponding intervertebral foramina) collectively constitute the cauda equina. Both the spinal cord (CNS) and spinal roots (PNS) are enveloped by meninges within the vertebral canal. Spinal nerves (which are formed in intervertebral foramina) are covered by connective tissue (epineurium, perineurium, & endoneurium) rather than meninges. -

CONGENITAL ABNORMALITIES of the CENTRAL NERVOUS SYSTEM Christopher Verity, Helen Firth, Charles Ffrench-Constant *I3
J Neurol Neurosurg Psychiatry: first published as 10.1136/jnnp.74.suppl_1.i3 on 1 March 2003. Downloaded from CONGENITAL ABNORMALITIES OF THE CENTRAL NERVOUS SYSTEM Christopher Verity, Helen Firth, Charles ffrench-Constant *i3 J Neurol Neurosurg Psychiatry 2003;74(Suppl I):i3–i8 dvances in genetics and molecular biology have led to a better understanding of the control of central nervous system (CNS) development. It is possible to classify CNS abnormalities Aaccording to the developmental stages at which they occur, as is shown below. The careful assessment of patients with these abnormalities is important in order to provide an accurate prog- nosis and genetic counselling. c NORMAL DEVELOPMENT OF THE CNS Before we review the various abnormalities that can affect the CNS, a brief overview of the normal development of the CNS is appropriate. c Induction—After development of the three cell layers of the early embryo (ectoderm, mesoderm, and endoderm), the underlying mesoderm (the “inducer”) sends signals to a region of the ecto- derm (the “induced tissue”), instructing it to develop into neural tissue. c Neural tube formation—The neural ectoderm folds to form a tube, which runs for most of the length of the embryo. c Regionalisation and specification—Specification of different regions and individual cells within the neural tube occurs in both the rostral/caudal and dorsal/ventral axis. The three basic regions of copyright. the CNS (forebrain, midbrain, and hindbrain) develop at the rostral end of the tube, with the spinal cord more caudally. Within the developing spinal cord specification of the different popu- lations of neural precursors (neural crest, sensory neurones, interneurones, glial cells, and motor neurones) is observed in progressively more ventral locations. -

Fetal MR How I Do It in Clinical Practice
Fetal MR How I do it in clinical practice M J Weston Leeds Why do it? • Add diagnostic certainty to US findings • Find additional anomalies • Research How is it done? • Trickier than you think. • Fast acquisition times • Signal to noise problems • Risk to fetus • T2 is mainstay • T1 useful for bowel and looking for fat or haemorrhage Cerebral MRI T2 weighted Other sequences T2 T1 DWI Change with time 24 weeks 36 weeks Spina bifida • MR is not a screening test • All cases detected by US • Confirmatory • Assignment of level • Visually very powerful for parents Spinal cord Conus Medullaris Spina bifida Head signs Spina bifida Sagittal US and MR are equally accurate at assigning level of lesion Aaronson OS et al. Radiology 2003; 227: 839‐843 Diastematomyelia Split cord Huge NTD on US Visual impact of MRI Caudal Regression Syndrome Caudal regression Head anomalies • Commonest indication – Apparently isolated ventriculomegaly • Establishing normal brain maturation • Problems with counselling… Unilateral hydrocephalus? • Near field reverberation Hydrocephalus Prognosis? Ventricular bleed Different sequences in bleed But, postnatally… Ultrasound Obstet Gynecol 2008; 32: 188 – 198 Good prognosis… Schizencephaly Artefact or schizencephaly? Follow-up Deep asymmetrical calcarine sulcus Arachnoid cyst Taiwan J Obstet Gynecol 2007; 46: 187 Prepontine arachnoid cyst Death of co-twin Obstet Gynecol 2011; 118: 928 – 940 Monochorionic – neurodevelopmental delay 26% of survivors Dichorionic ‐ 2% 2 weeks later Microcephaly etc Face and holoprosencephaly Face and head Facial cleft and no eyes Normal Sent for head but… But also has small lungs Problem solving Fetal kidneys Inclusion cyst Cervical teratoma Neck lymphangioma Nasopharyngeal teratoma? Focal bulge What is this? Co‐existant Mole Retroplacental bleed Intrapartum scar rupture Conclusions • Complimentary to US • Added worth is less if expert US • Prognostic difficulties • Changing the role of the Radiologist. -

Classification of Congenital Abnormalities of the CNS
315 Classification of Congenital Abnormalities of the CNS M. S. van der Knaap1 A classification of congenital cerebral, cerebellar, and spinal malformations is pre J . Valk2 sented with a view to its practical application in neuroradiology. The classification is based on the MR appearance of the morphologic abnormalities, arranged according to the embryologic time the derangement occurred. The normal embryology of the brain is briefly reviewed, and comments are made to explain the classification. MR images illustrating each subset of abnormalities are presented. During the last few years, MR imaging has proved to be a diagnostic tool of major importance in children with congenital malformations of the eNS [1]. The excellent gray fwhite-matter differentiation and multi planar imaging capabilities of MR allow a systematic analysis of the condition of the brain in infants and children. This is of interest for estimating prognosis and for genetic counseling. A classification is needed to serve as a guide to the great diversity of morphologic abnormalities and to make the acquired data useful. Such a system facilitates encoding, storage, and computer processing of data. We present a practical classification of congenital cerebral , cerebellar, and spinal malformations. Our classification is based on the morphologic abnormalities shown by MR and on the time at which the derangement of neural development occurred. A classification based on etiology is not as valuable because the various presumed causes rarely lead to a specific pattern of malformations. The abnor malities reflect the time the noxious agent interfered with neural development, rather than the nature of the noxious agent. The vulnerability of the various structures to adverse agents is greatest during the period of most active growth and development. -

The Conus Medullaris: a Comprehensive Review
THE SPINE SCHOLAR VOLUME 1, NUMBER 2, 2017 SEATTLE SCIENCE FOUNDATION REVIEW The Conus Medullaris: A Comprehensive Review Garrett Ng1, Anthony V. D’Antoni1, R. Shane Tubbs2 1 The CUNY School of Medicine, New York, NY 10031, USA 2 Department of Anatomical Sciences, St. George’s University, Grenada http:thespinescholar.com https:doi.org/10.26632/ss.10.2017.1.2 Key Words: anatomy, embryology, spinal cord, spine ABSTRACT The position of the conus medullaris within the vertebral canal varies. Given its role in sensory and motor function, a comprehensive understanding of the conus medullaris is necessary. PubMed and Google Scholar were used to review the literature on the conus medullaris. Pathological states and traumatic injury relating to the conus medullaris should be studied further. Spine Scholar 1:93-96, 2017 INTRODUCTION The conus medullaris (Fig. 1), also known as the medullary cone, is the distal end of the spinal cord. Its location varies, and in adults it tapers at approximately the first or second lumbar vertebra, ranging from T11 and L3 (Neel, 2016). Derived from the neural tube, the structure ascends in the vertebral canal because the growth rates of the spinal cord and the vertebral column differ during development (Salbacak et al., 2000). Figure 1: Schematic drawing of the conus medullaris and distal nerve roots in relation to the sacrum. The conus contains the sacral and coccygeal segments of the spinal cord (Taylor and Coolican, 1988). Criteria for recognizing the conus on computed tomography (CT) scans have been published by Grogan et al. (1984). The radiological properties of the structure have been studied through magnetic resonance imaging (MRI) (Saifuddin et al., 1997). -

Spinal Lipoma Associated with Congenital Dermal Sinus: a Case Report
Rios LTM et al. LipomaRELATO espinhal DEassociado CASO •a CASEseio dérmico REPORT congênito Lipoma espinhal associado a seio dérmico congênito: relato de caso* Spinal lipoma associated with congenital dermal sinus: a case report Lívia Teresa Moreira Rios1, Ricardo Villar Barbosa de Oliveira2, Marília da Glória Martins3, Olga Maria Ribeiro Leitão4, Vanda Maria Ferreira Simões5, Janilson Moucherek Soares do Nascimento6 Resumo Os lipomas espinhais são raros, respondendo por 1% de todos os tumores espinhais, estando associados ao disra- fismo espinhal oculto em mais de 99% dos casos. Estão divididos em três tipos principais: lipomielomeningocele, lipoma intradural e fibrolipoma do filo terminal. Este relato descreve um caso de lipoma lombossacral congênito asso- ciado a estigma cutâneo do tipo seio dérmico lombar congênito. Unitermos: Disrafismo espinhal oculto; Seio dérmico congênito; Lipoma intradural; Ultrassonografia. Abstract Spinal lipomas are rare, accounting for 1% of all spinal tumors and being associated with occult spinal dysraphism in more than 99% of cases. Such lesions are divided into three main types, namely, lipomyelomeningoceles, intradural lipomas, and filum terminale fibrolipomas. The present report describes a case of congenital lumbosacral lipoma associated with cutaneous stigmata of the lumbar dermal sinus type. Keywords: Occult spinal dysraphism; Congenital dermal sinus; Intradural lipoma; Ultrasonography. Rios LTM, Oliveira RVB, Martins MG, Leitão OMR, Simões VMF, Nascimento JMS. Lipoma espinhal associado a seio dérmico congê- nito: relato de caso. Radiol Bras. 2011 Jul/Ago;44(4):265–267. INTRODUÇÃO tigma cutâneo associado, do tipo massa compatível com seio dérmico dorsal. Des- coberta de pele, tufo de cabelo, apêndice tacava-se ainda pequena massa sólida re- Os disrafismos espinhais ocultos são cutâneo, pele com distúrbio de coloração, coberta por pele na região lombossacra um grupo de afecções dorsais que existem ou depressão cutânea(1–3). -

227 INTRODUCTION: Diastematomyelia (Also Known As a Split Cord Malformation) Is a Rare Dysraphic Lesion of the Spinal Cord in W
Role of rehabilitation a case of diastematomyelia Stanescu Ioana¹, Kallo Rita¹, Bulboaca Adriana³, Dogaru Gabriela ² 1.Rehabilitation Hospital Cluj, Neurology Department 2.Rehabilitation Hospital Cluj, Physical Medicine and Rehabilitation Department 3.University of Medecine and Pharmacy Cluj - Physiopathology Department Balneo Research Journal DOI: http://dx.doi.org/10.12680/balneo.2017.156 Vol.8, No.4, December 2017 p: 227 – 230 Corresponding author: Gabriela Dogaru, E-mail address: [email protected] Abstract Diastematomyelia (split cord malformation) is a rare dysraphic lesion in which a part of the spinal cord is split in the sagittal plane into two hemicords, a bony, cartilagenous or fibrous spur projecting through the dura mater is visible in 33% of cases. Vertebral anomalies (spina bifida) are common. It occurs usually between D9 and S1. Classificatin includes two types: type 1 with a duplicated dural sac, with common midline spur, usually symptomatic, and type 2 with a single dural sac and usually less symptomatic. The majority of patients are presenting with tethered cord syndrome (neurologic deficits in the lower limbs and perineum). MRI is the modality of choice for diagnosis. In symptomatic cases, surgical release of the cord and resection of spur with repair of dura are performed, with good results. We present a case of a pauci-symptomatic type 1 dyastematomyelia, manifested by intermittent and resistant lumbar pain, in which physiotherapy during rehabilitation program have shown to improve pain intensity. Key words: spinal cord malformation, dyastematomyelia, lumbar spine, INTRODUCTION: Diastematomyelia (also Intramedullary tumours associated with known as a split cord malformation) is a rare diastematomyelia have been rarely described and dysraphic lesion of the spinal cord in which a part associated conditions like a tethered cord, inclusion of the spinal cord is split in the sagittal plane into dermoid, lipoma, syringo-hydromyelia and Chiari two hemicords. -

Diastematomyelia: a Case with Familial Aggregation of Neural Tube Defects
Case Study TheScientificWorldJOURNAL (2004) 4, 847–852 ISSN 1537-744X; DOI 10.1100/tsw.2004.140 Diastematomyelia: A Case with Familial Aggregation of Neural Tube Defects Nuray Öksüz Kanbur1,*, Pınar Güner2, Orhan Derman1, Nejat Akalan3, Ayşenur Cila4, and Tezer Kutluk1 1Section of Adolescent Medicine, Department of Pediatrics, Hacettepe University Faculty of Medicine, 06100 Ankara, Turkey; 2Department of Public Health, Hacettepe University Faculty of Medicine, Ankara, Turkey; 3Department of Neurosurgery, Hacettepe University Faculty of Medicine, Ankara, Turkey; 4Department of Radiology, Hacettepe University Faculty of Medicine, Ankara, Turkey E-mail: [email protected] Received July 2, 2004; Revised August 31, 2004; Accepted August 31, 2004; Published September 21, 2004 Intrauterine neural tube defects, meningomyelocele, and diastematomyelia are developmental errors at different stages of the closure of the neural tube. The familial aggregation of these neural tube defects is not previously reported in the literature and should make one think about a common embryogenesis and a possible common mechanism of etiopathogenesis leading to anomalies at different stages of this embryogenesis. This paper presents a 12-year-old Turkish boy with diastematomyelia who was suspected with a demonstrative dermatologic finding without any neurologic sign and diagnosed with magnetic resonance imaging (MRI). He has a positive family history of a stillbirth with neural tube defect, an exitus with meningomyelocele, and an epileptic child in his female siblings. KEYWORDS: diastematomyelia, neural tube defect, familial aggregation, genetics, pediatrics, Turkey DOMAINS: child health and human development, medical care, nursing INTRODUCTION Diastematomyelia is a form of spinal dysraphism, defined as sagittal division of the spinal cord into two hemicords, each of which contains a central canal. -
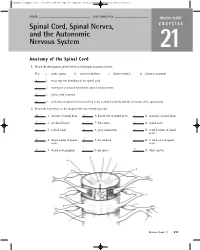
Spinal Cord, Spinal Nerves, and the Autonomic Nervous System
ighapmLre21pg211_216 5/12/04 2:24 PM Page 211 impos03 302:bjighapmL:ighapmLrevshts:layouts: NAME ___________________________________ LAB TIME/DATE _______________________ REVIEW SHEET Spinal Cord, Spinal Nerves, exercise and the Autonomic Nervous System 21 Anatomy of the Spinal Cord 1. Match the descriptions given below to the proper anatomical term: Key: a. cauda equina b. conus medullaris c. filum terminale d. foramen magnum d 1. most superior boundary of the spinal cord c 2. meningeal extension beyond the spinal cord terminus b 3. spinal cord terminus a 4. collection of spinal nerves traveling in the vertebral canal below the terminus of the spinal cord 2. Match the key letters on the diagram with the following terms. m 1. anterior (ventral) hornn 6. dorsal root of spinal nervec 11. posterior (dorsal) horn k 2. arachnoid materj 7. dura materf 12. spinal nerve a 3. central canalo 8. gray commissure i 13. ventral ramus of spinal nerve h 4. dorsal ramus of spinald 9. lateral horne 14. ventral root of spinal nerve nerve g l 5. dorsal root ganglion 10. pia materb 15. white matter o a b n c m d e l f g k h j i Review Sheet 21 211 ighapmLre21pg211_216 5/12/04 2:24 PM Page 212 impos03 302:bjighapmL:ighapmLrevshts:layouts: 3. Choose the proper answer from the following key to respond to the descriptions relating to spinal cord anatomy. Key: a. afferent b. efferent c. both afferent and efferent d. association d 1. neuron type found in posterior hornb 4. fiber type in ventral root b 2.