Silicon, Germanium and Tin Arsines
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

MDA Scientific SPM
MDA Scientific SPM Fast response monitor for the detection of a target gas SPM Advantages • Fast response monitor specific to target gas only • Gas sensitivity to ppb levels with physical evidence • Minimum maintenance and no dynamic calibration • Customized for harsh industrial environments • More than 50 gas calibrations available Applications • Outdoor locations • Corrosive areas • Remote sampling areas • Gas storage areas • Survey work • Perimeter/fencelines • Ventilation and exhaust systems Options • Z-purge system • Duty cycle • ChemKey™ • RS422 • Remote reset • Portable • Extended sample • Heater option (operate from -20°C to ±40°C) Technical Specification Specifications Detection Technique Chemcassette® Detection System Alarm Point Dual level alarms typically set at 1/2 TLV and TLV Response Time As fast as 10 seconds Alarm Indication Local audio/visual alarms; remote capability optional Signal Outputs SPDT concentration alarm relays; SPDT fault relay; 4-20 mA; digital display Relay Rating 120VAC@10amps; 240VAC@5amps; 48VDC@5amps Operating Temperature Range 32° to 104°F; 0° to 40°C (basic unit); heating/cooling optional Power Requirements 115/230 VAC 50/60 Hz, battery operation optional Enclosure NEMA 4X fiberglass (basic unit) Dimensions 12"(H) x 12"(W) x 7”(D) (30.5 x 30.5 x 17.8 cm) (basic unit) Weight Up to 25 pounds, depending on option installed Note: options may vary the specifications Detectable Gases Range Amines Ammonia (NH3) 2.6-75.0 ppm Ammonia (NH3)-II 2.6-75.0 ppm Dimethylamine (DMA) 0.1-6 ppm n-Butylamine (n-BA) 0.4-12 -

Vibrationally Excited Hydrogen Halides : a Bibliography On
VI NBS SPECIAL PUBLICATION 392 J U.S. DEPARTMENT OF COMMERCE / National Bureau of Standards National Bureau of Standards Bldg. Library, _ E-01 Admin. OCT 1 1981 191023 / oO Vibrationally Excited Hydrogen Halides: A Bibliography on Chemical Kinetics of Chemiexcitation and Energy Transfer Processes (1958 through 1973) QC 100 • 1X57 no. 2te c l !14 c '- — | NATIONAL BUREAU OF STANDARDS The National Bureau of Standards' was established by an act of Congress March 3, 1901. The Bureau's overall goal is to strengthen and advance the Nation's science and technology and facilitate their effective application for public benefit. To this end, the Bureau conducts research and provides: (1) a basis for the Nation's physical measurement system, (2) scientific and technological services for industry and government, (3) a technical basis for equity in trade, and (4) technical services to promote public safety. The Bureau consists of the Institute for Basic Standards, the Institute for Materials Research, the Institute for Applied Technology, the Institute for Computer Sciences and Technology, and the Office for Information Programs. THE INSTITUTE FOR BASIC STANDARDS provides the central basis within the United States of a complete and consistent system of physical measurement; coordinates that system with measurement systems of other nations; and furnishes essential services leading to accurate and uniform physical measurements throughout the Nation's scientific community, industry, and commerce. The Institute consists of a Center for Radiation Research, an Office of Meas- urement Services and the following divisions: Applied Mathematics — Electricity — Mechanics — Heat — Optical Physics — Nuclear Sciences" — Applied Radiation 2 — Quantum Electronics 1 — Electromagnetics 3 — Time 3 1 1 and Frequency — Laboratory Astrophysics — Cryogenics . -

Thursday 10 January 2019
Please check the examination details below before entering your candidate information Candidate surname Other names Pearson Edexcel Centre Number Candidate Number International Advanced Level Thursday 10 January 2019 Afternoon (Time: 1 hour 40 minutes) Paper Reference WCH04/01 Chemistry Advanced Unit 4: General Principles of Chemistry I – Rates, Equilibria and Further Organic Chemistry (including synoptic assessment) Candidates must have: Scientific calculator Total Marks Data Booklet Instructions • Use black ink or black ball-point pen. • Fill in the boxes at the top of this page with your name, centre number and candidate number. • Answer all questions. • Answer the questions in the spaces provided – there may be more space than you need. Information • The total mark for this paper is 90. • The marks for each question are shown in brackets – use this as a guide as to how much time to spend on each question. • Questions labelled with an asterisk (*) are ones where the quality of your written communication will be assessed – you should take particular care with your spelling, punctuation and grammar, as well as the clarity of expression, on these questions. • A Periodic Table is printed on the back cover of this paper. Advice • Read each question carefully before you start to answer it. • Show all your working in calculations and include units where appropriate. • Check your answers if you have time at the end. Turn over P54560A ©2019 Pearson Education Ltd. *P54560A0128* 2/1/1/1/1/1/ SECTION A Answer ALL the questions in this section. You should aim to spend no more than 20 minutes on THIS AREA WRITE IN DO NOT this section. -

The Summer Assignment Will Receive a GRADE on the First Day of Class – August 9
Bishop Moore AP Chemistry Summer Assignment June 2017 Future AP Chemistry Student, Welcome to AP Chemistry. In order to ensure the best start for everyone next fall, I have prepared a summer assignment that reviews basic chemistry concepts some of which you may have forgotten you learned. For those topics you need help with there are a multitude of tremendous chemistry resources available on the Internet. With access to hundreds of websites either in your home or at the local library, I am confident that you will have sufficient resources to prepare adequately for the fall semester. The reference text book as part of AP course is “Chemistry: The Central Science” by Brown LeMay 14th Edition for AP. Much of the material in this summer packet will be familiar to you. It will be important for everyone to come to class the first day prepared. While I review throughout the course, extensive remediation is not an option as we work towards our goal of being 100% prepared for the AP Exam in early May. There will be a test covering the basic concepts included in the summer packet during the first or second week of school. You may contact me by email: ([email protected]) this summer. I will do my best to answer your questions ASAP. I hope you are looking forward to an exciting year of chemistry. You are all certainly excellent students, and with plenty of motivation and hard work you should find AP Chemistry a successful and rewarding experience. Finally, I recommend that you spread out the summer assignment. -

IODINE Its Properties and Technical Applications
IODINE Its Properties and Technical Applications CHILEAN IODINE EDUCATIONAL BUREAU, INC. 120 Broadway, New York 5, New York IODINE Its Properties and Technical Applications ¡¡iiHiüíiüüiütitittüHiiUitítHiiiittiíU CHILEAN IODINE EDUCATIONAL BUREAU, INC. 120 Broadway, New York 5, New York 1951 Copyright, 1951, by Chilean Iodine Educational Bureau, Inc. Printed in U.S.A. Contents Page Foreword v I—Chemistry of Iodine and Its Compounds 1 A Short History of Iodine 1 The Occurrence and Production of Iodine ....... 3 The Properties of Iodine 4 Solid Iodine 4 Liquid Iodine 5 Iodine Vapor and Gas 6 Chemical Properties 6 Inorganic Compounds of Iodine 8 Compounds of Electropositive Iodine 8 Compounds with Other Halogens 8 The Polyhalides 9 Hydrogen Iodide 1,0 Inorganic Iodides 10 Physical Properties 10 Chemical Properties 12 Complex Iodides .13 The Oxides of Iodine . 14 Iodic Acid and the Iodates 15 Periodic Acid and the Periodates 15 Reactions of Iodine and Its Inorganic Compounds With Organic Compounds 17 Iodine . 17 Iodine Halides 18 Hydrogen Iodide 19 Inorganic Iodides 19 Periodic and Iodic Acids 21 The Organic Iodo Compounds 22 Organic Compounds of Polyvalent Iodine 25 The lodoso Compounds 25 The Iodoxy Compounds 26 The Iodyl Compounds 26 The Iodonium Salts 27 Heterocyclic Iodine Compounds 30 Bibliography 31 II—Applications of Iodine and Its Compounds 35 Iodine in Organic Chemistry 35 Iodine and Its Compounds at Catalysts 35 Exchange Catalysis 35 Halogenation 38 Isomerization 38 Dehydration 39 III Page Acylation 41 Carbón Monoxide (and Nitric Oxide) Additions ... 42 Reactions with Oxygen 42 Homogeneous Pyrolysis 43 Iodine as an Inhibitor 44 Other Applications 44 Iodine and Its Compounds as Process Reagents ... -

Gas Phase Chemistry and Removal of CH3I During a Severe Accident
DK0100070 Nordisk kernesikkerhedsforskning Norraenar kjarnoryggisrannsoknir Pohjoismainenydinturvallisuustutkimus Nordiskkjernesikkerhetsforskning Nordisk karnsakerhetsforskning Nordic nuclear safety research NKS-25 ISBN 87-7893-076-6 Gas Phase Chemistry and Removal of CH I during a Severe Accident Anna Karhu VTT Energy, Finland 2/42 January 2001 Abstract The purpose of this literature review was to gather valuable information on the behavior of methyl iodide on the gas phase during a severe accident. The po- tential of transition metals, especially silver and copper, to remove organic io- dides from the gas streams was also studied. Transition metals are one of the most interesting groups in the contex of iodine mitigation. For example silver is known to react intensively with iodine compounds. Silver is also relatively inert material and it is thermally stable. Copper is known to react with some radioio- dine species. However, it is not reactive toward methyl iodide. In addition, it is oxidized to copper oxide under atmospheric conditions. This may limit the in- dustrial use of copper. Key words Methyl iodide, gas phase, severe accident mitigation NKS-25 ISBN 87-7893-076-6 Danka Services International, DSI, 2001 The report can be obtained from NKS Secretariat P.O. Box 30 DK-4000RoskiIde Denmark Phone +45 4677 4045 Fax +45 4677 4046 http://www.nks.org e-mail: [email protected] Gas Phase Chemistry and Removal of CH3I during a Severe Accident VTT Energy, Finland Anna Karhu Abstract The purpose of this literature review was to gather valuable information on the behavior of methyl iodide on the gas phase during a severe accident. -

Production Process for Refined Hydrogen Iodide
^ ^ ^ ^ I ^ ^ ^ ^ ^ ^ II ^ II ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ ^ I ^ European Patent Office Office europeen des brevets EP 0 714 849 A1 EUROPEAN PATENT APPLICATION (43) Date of publication: (51) |nt CI.6: C01B7/13 05.06.1996 Bulletin 1996/23 (21) Application number: 95308524.8 (22) Date of filing: 28.11.1995 (84) Designated Contracting States: • Tanaka, Yoshinori DE FR GB NL Mobara-shi, Chiba (JP) • Omura, Masahiro (30) Priority: 28.11.1994 J P 292771/95 Mobara-shi, Chiba (JP) • Tomoshige, Naoki (71) Applicant: MITSUI TOATSU CHEMICALS, Mobara-shi, Chiba (JP) INCORPORATED Tokyo (JP) (74) Representative: Nicholls, Kathryn Margaret et al MEWBURN ELLIS (72) Inventors: York House • Utsunomiya, Atsushi 23 Kingsway Takaishi-shi, Osaka (JP) London WC2B 6HP (GB) • Sasaki, Kenju Mobara-shi, Chiba (JP) (54) Production process for refined hydrogen iodide (57) A process for producing refined hydrogen io- amount of which is at least 1/3 (weight ratio) relative to dide by contacting crude hydrogen iodide with a zeolite the amount of the zeolite, to convert impurities of sulfur is disclosed. Crude hydrogen iodide is obtained by re- components contained in the zeolite to hydrogen ducing iodine with a hydrogenated naphthalene, where- sulfide, thereby removing the sulfur components. The in all of iodine is dissolved in advance in a part of the kind of the zeolite is an A type zeolite (an average pore hydrogenated naphthalene to prepare an iodine solu- diameter: 3 angstrom, 4 angstrom or 5 angstrom) or a tion, and the reaction is carried out while adding contin- mordenite type zeolite. Further, an activated carbon uously or intermittently the above solution to the balance may be combined with the zeolite at the rear stage there- of the hydrogenated naphthalene; and the same oper- of. -

Chapter 9 Hydrogen
Chapter 9 Hydrogen Diborane, B2 H6• is the simplest member of a large class of compounds, the electron-deficient boron hydrid Like all boron hydrides, it has a positive standard free energy of formation, and so cannot be prepared direct from boron and hydrogen. The bridge B-H bonds are longer and weaker than the tenninal B-H bonds (13: vs. 1.19 A). 89.1 Reactions of hydrogen compounds? (a) Ca(s) + H2Cg) ~ CaH2Cs). This is the reaction of an active s-me: with hydrogen, which is the way that saline metal hydrides are prepared. (b) NH3(g) + BF)(g) ~ H)N-BF)(g). This is the reaction of a Lewis base and a Lewis acid. The product I Lewis acid-base complex. (c) LiOH(s) + H2(g) ~ NR. Although dihydrogen can behave as an oxidant (e.g., with Li to form LiH) or reductant (e.g., with O2 to form H20), it does not behave as a Br0nsted or Lewis acid or base. It does not r with strong bases, like LiOH, or with strong acids. 89.2 A procedure for making Et3MeSn? A possible procedure is as follows: ~ 2Et)SnH + 2Na 2Na+Et3Sn- + H2 Ja'Et3Sn- + CH3Br ~ Et3MeSn + NaBr 9.1 Where does Hydrogen fit in the periodic chart? (a) Hydrogen in group 1? Hydrogen has one vale electron like the group 1 metals and is stable as W, especially in aqueous media. The other group 1 m have one valence electron and are quite stable as ~ cations in solution and in the solid state as simple 1 salts. -
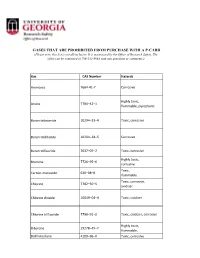
Lists of Gases for Pcard Manual.Xlsx
GASES THAT ARE PROHIBITED FROM PURCHASE WITH A P-CARD (Please note, this list is not all-inclusive. It is maintained by the Office of Research Safety. The office can be contacted at 706-542-9088 with any questions or comments.) Gas CAS Number Hazards Ammonia 7664‐41‐7 Corrosive Highly toxic, Arsine 7784–42–1 flammable, pyrophoric Boron tribromide 10294–33–4 Toxic, corrosive Boron trichloride 10294–34–5 Corrosive Boron trifluoride 7637–07–2 Toxic, corrosive Highly toxic, Bromine 7726–95–6 corrosive Toxic, Carbon monoxide 630–08–0 flammable Toxic, corrosive, Chlorine 7782–50–5 oxidizer Chlorine dioxide 10049–04–4 Toxic, oxidizer Chlorine trifluoride 7790–91–2 Toxic, oxidizer, corrosive Highly toxic, Diborane 19278–45–7 flammable, Dichlorosilane 4109–96–0 Toxic, corrosive Ethylene oxide 75–21–8 Toxic, flammable Highly toxic, corrosive, Fluorine 7782–41–4 oxidizer Highly toxic, Germane 7782–65–2 flammable Hydrogen bromide 10035–10–6 Toxic, corrosive Hydrogen chloride 7647–01–0 Toxic, corrosive Hydrogen cyanide 74–90–8 Highly toxic, flammable Hydrogen fluoride 7664–39–3 Toxic, corrosive Hydrogen iodide 10034‐85‐2 Toxic, corrosive Highly toxic, Hydrogen selenide 7783–07–5 flammable Toxic, flammable, Hydrogen sulfide 7783–06–4 corrosive Methyl bromide 74–83–9 Toxic Methyl isocyanate 624‐83‐9 Highly toxic, flammable Methyl mercaptan 74–93–1 Toxic, flammable Nickel carbonyl 13463–39–3 Highly toxic, flammable Highly toxic, Nitric oxide 10102–43–9 oxidizer Highly toxic, Nitrogen dioxide 10102–44–0 oxidizer, corrosive Highly toxic, Ozone 10028‐15‐6 -

Signature Redacted 0'10
A STUDY OF THE REACTION 0'10 OF HYDROGEN IODIDE WITH SILICON v193 ARY THESIS by CAROLYN H. KLEIN S.B. Simmons College, 1933. Submitted in Partial Fulfillment of the Requirements for the Degree of MASTER OF SCIENCE from the Massachusetts Institute of Technology 1934 Signature Redacted Signature of Author. .. .. Department of Chemistry. Date Professor in Charge of Research . Signature Redacted Chairman of Departmental Commltee on Graduate Students . Signature Rected Signature Redacted Head of Deartment . it * - Acknowledgement The author wishes to express her most sincere appreciation to Professor Walter C. Schumb for the active interest which he has at all times shown in this research, and for the many helpful suggestions which he has offered. The author should also like to extend her thanks to the many other members of the Research Laboratory of Inorganic Chemistry, for the helpful advice which they have given. N I 197618 K j\> Table of Contents Page I Previous Wiork on the Preparation of the Iodides of Silicon . .... .. 1 II Introauction .. .... ..... 3 III Preparation of Hydrogen Iodide . 4 IV Figure I. Apparatus Used in the Reaction between Silicon and HI .. ... .. 6 V The Reaction of Silicon withHI ....... 7 VI Figure II. Revised Apparatus for the Reaction of Silicon with HI . .. .10 VII General Conclusions from the Reaction between Silicon and HI ..... .... 13 VIII Reaction of Calcium Silicide with HI .. ... 14 IX Figure III. Arparatus Used for the Reaction of Calcium Silicide with HI .. 15 X Figure IV. Apparatus for Distillation of the Crude Product of the Reaction .. 19 XI The Purification of Silicon Tetra-iodide .. -

Safety Data Sheet
SAFETY DATA SHEET Revision Date 14-Feb-2020 Revision Number 2 1. Identification Product Name Arsenic (III) iodide Cat No. : 45067 CAS-No 7784-45-4 Synonyms Arsenic triiodide. Recommended Use Laboratory chemicals. Uses advised against Food, drug, pesticide or biocidal product use. Details of the supplier of the safety data sheet Company Alfa Aesar Thermo Fisher Scientific Chemicals, Inc. 30 Bond Street Ward Hill, MA 01835-8099 Tel: 800-343-0660 Fax: 800-322-4757 Email: [email protected] www.alfa.com Emergency Telephone Number During normal business hours (Monday-Friday, 8am-7pm EST), call (800) 343-0660. After normal business hours, call Carechem 24 at (866) 928-0789. 2. Hazard(s) identification Classification This chemical is considered hazardous by the 2012 OSHA Hazard Communication Standard (29 CFR 1910.1200) Acute oral toxicity Category 3 Acute Inhalation Toxicity - Dusts and Mists Category 3 Carcinogenicity Category 1A Label Elements Signal Word Danger Hazard Statements May cause cancer Toxic if swallowed or if inhaled ______________________________________________________________________________________________ Page 1 / 7 Arsenic (III) iodide Revision Date 14-Feb-2020 ______________________________________________________________________________________________ Precautionary Statements Prevention Obtain special instructions before use Do not handle until all safety precautions have been read and understood Use personal protective equipment as required Wash face, hands and any exposed skin thoroughly after handling Do not eat, drink or smoke when using this product Avoid breathing dust/fume/gas/mist/vapors/spray Use only outdoors or in a well-ventilated area Response IF exposed or concerned: Get medical attention/advice Inhalation IF INHALED: Remove victim to fresh air and keep at rest in a position comfortable for breathing Call a POISON CENTER or doctor/physician Ingestion IF SWALLOWED: Immediately call a POISON CENTER or doctor/physician Rinse mouth Storage Store locked up Store in a well-ventilated place. -

Thesis by Robert T. Dillon and William G. Young in Partial Fulfillment of The
RESE.i\RCHES ON THE NOnM.AL BUTENES Thesis by Robert T. Dillon and William G. Young In partial fulfillment of the requirements for the degree of Doctor of Philosophy California Institute of Technology Pasadena, California 1929 TABLE OF CONTBNTS 1. Acknowledgments. 2. The Synthesis of 1-Butene. Robert T. Dillon 3. The preparation of .Anhydrous Hydrogen Iodide. Robert T. Dillon and 'iifilliam G. Young. 4. The Synthesis of the Isomeric 2-Butenes. William G. Young and hobert T. Dillon 5. The Condensation of Acetaldehyde with Methylmalonic Ester: Methylations with Methyl Bromide. William G. Young 6. The Reaction Rates of Potassium Iodide with 1,2- and 2,3-Dibromo butane and its Application to the Analysis of Mixtures of the Nonnal Butene s. Robert T. Dillon and William G. Young 7. The Probable Mechanism of the Reaction of AlSylene Bromides with Potassium Iodide. Robert T. Dillon. Acknowledgments The authors wish to express their deep appreciation to Professor Howard J. Lucas for his guidance, advice and counsel in the work involved in these researches. They also wish to thank Mrs. A.M.Morrill, Mr. S.E.Parker, Mr. E.H.Searle, and other members of the d~partment, who have cooperated in every way. The first, second, third and fifth papers contain results obtained in an investigation listed as Project 14 of the .American Petroleum Institute Research. Financial assistance in this work has been received from a research fund of the American Petroleum Institute donated by Mr. John D. Rockefeller. This fund was ad- ministered by the Institute with the cooperation of the Central "' Petroleum.