Synthesis of Cysteine Sulfoxides and Related Compounds Occurring in Wild Onions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

First Insights Into the Mode of Action of a "Lachrymatory Factor Synthase"
Phytochemistry 72 (2011) 1939–1946 Contents lists available at ScienceDirect Phytochemistry journal homepage: www.elsevier.com/locate/phytochem First insights into the mode of action of a ‘‘lachrymatory factor synthase’’ – Implications for the mechanism of lachrymator formation in Petiveria alliacea, Allium cepa and Nectaroscordum species ⇑ Quan He a, Roman Kubec b, Abhijit P. Jadhav a, Rabi A. Musah a, a Department of Chemistry, University at Albany, State University of New York, Albany, NY 12222, USA b Department of Applied Chemistry, University of South Bohemia, Branišovská 31, 370 05 Cˇeské Budeˇjovice, Czech Republic article info abstract Article history: A study of an enzyme that reacts with the sulfenic acid produced by the alliinase in Petiveria alliacea L. Received 16 December 2010 (Phytolaccaceae) to yield the P. alliacea lachrymator (phenylmethanethial S-oxide) showed the protein Received in revised form 11 July 2011 to be a dehydrogenase. It functions by abstracting hydride from sulfenic acids of appropriate structure Available online 15 August 2011 to form their corresponding sulfines. Successful hydride abstraction is dependent upon the presence of a benzyl group on the sulfur to stabilize the intermediate formed on abstraction of hydride. This dehy- Keywords: drogenase activity contrasts with that of the lachrymatory factor synthase (LFS) found in onion, which Petiveria alliacea catalyzes the rearrangement of 1-propenesulfenic acid to (Z)-propanethial S-oxide, the onion lachryma- Phytolaccaceae tor. Based on the type of reaction it catalyzes, the onion LFS should be classified as an isomerase and Lachrymatory factor synthase Sulfenic acid would be called a ‘‘sulfenic acid isomerase’’, whereas the P. alliacea LFS would be termed a ‘‘sulfenic acid Sulfenic acid dehydrogenase dehydrogenase’’. -

Allium Macleanii Baker, 1883
Allium macleanii Baker, 1883 Identifiants : 1573/allmac Association du Potager de mes/nos Rêves (https://lepotager-demesreves.fr) Fiche réalisée par Patrick Le Ménahèze Dernière modification le 30/09/2021 Classification phylogénétique : Clade : Angiospermes ; Clade : Monocotylédones ; Ordre : Asparagales ; Famille : Amaryllidaceae ; Classification/taxinomie traditionnelle : Règne : Plantae ; Sous-règne : Tracheobionta ; Division : Magnoliophyta ; Classe : Liliopsida ; Ordre : Liliales ; Famille : Amaryllidaceae ; Sous-famille : Allioideae ; Tribu : Allieae ; Genre : Allium ; Synonymes : x (=) basionym, Allium elatum Regel 1884 ; Nom(s) anglais, local(aux) et/ou international(aux) : Oriental royal Salep, royal salep , kabullök (sv) ; Note comestibilité : *** Rapport de consommation et comestibilité/consommabilité inférée (partie(s) utilisable(s) et usage(s) alimentaire(s) correspondant(s)) : Racine (bulbes : crus ou cuits [nourriture(dp*)µ/alimentµ27(+x) et/ou assaisonnement : aromate (fines-herbes et/ou{{{(dp*) condiment27(+x) aromatique{{{(dp*))]) et feuille (feuilles : idem bulbes|{{{(dp*)(27(+x)), ex. comme potherbe{{{(dp*)) comestibles.(1*) Les bulbes sont consommés crus ou cuits. Les feuilles sont consommées crues ou cuites. Les fleurs sont utilisées crues pour parfumer les salades Partie testée : bulbe{{{0(+x) (traduction automatique) Original : Bulb{{{0(+x) Taux d'humidité Énergie (kj) Énergie (kcal) Protéines (g) Pro- Vitamines C (mg) Fer (mg) Zinc (mg) vitamines A (µg) 0 0 0 0 0 0 0 (1*)Voir genre Allium pour les précautions à prendre (risques de confusion et possible toxicité à fortes doses).(1*)Voir genre Allium pour les précautions à prendre (risques de confusion et possible toxicité à fortes doses){{{(rp*). Note médicinale : ** Illustration(s) (photographie(s) et/ou dessin(s)): Page 1/3 Autres infos : Plante importante localement{{{27(+x). -

(Curculigo Orchioides) and Salam (Eulophia Compestris) Madan B
Int J Ayu Pharm Chem REVIEW ARTICLE www.ijapc.com e-ISSN 2350-0204 A Review Article on Species used as Musali (Curculigo orchioides) and Salam (Eulophia compestris) Madan B. Tonge* *Dept. of Dravyaguna, Govt. Ayurved College Nanded (MS), India Abstract In day to day practice when we see the market samples of Musali it creates confusion in mind; which type Musali is sold by the vendor. These days various species of plants are used as Musali in different parts of India. Traditionally, Salam and Salam panja are also used as Mushali. To rule out all these differences and arrive to a definite conclusion. This is an attempt to collect the referances from samhitas and nighantus about musali. Botanically classify the species which are used as musali. Describe all the species which are in use as musali in a systematic manner. Keywords Mushali, Shweta Musali, Salam, Talmuli Greentree Group Received 09/08/16 Accepted 29/08/16 Published 10/09/16 ________________________________________________________________________________________________________ Madan B Tonge 2016 Greentree Group © IJAPC Int J Ayu Pharm Chem 2016 Vol. 5 Issue 2 www.ijapc.com 283 [e ISSN 2350-0204] Int J Ayu Pharm Chem INTRODUCTION shwveta musali few species of asparagaceae The term musali is famous in traditional family are in use and also roots of salam Indian system of medicine. Medicine with mishri and salampanja mishri are used as musali name is known to many household in musali. The word mishri is derived from India. Most commonly used as a tonic, musali, so few people call it as salam aphrodisiac, rejuvenator for increasing musali, salam panja musali. -

The Republic of Tajikistan Ministry of Energy and Industry
The Republic of Tajikistan Ministry of Energy and Industry DATA COLLECTION SURVEY ON THE INSTALLMENT OF SMALL HYDROPOWER STATIONS FOR THE COMMUNITIES OF KHATLON OBLAST IN THE REPUBLIC OF TAJIKISTAN FINAL REPORT September 2012 Japan International Cooperation Agency NEWJEC Inc. E C C CR (1) 12-005 Final Report Contents, List of Figures, Abbreviations Data Collection Survey on the Installment of Small Hydropower Stations for the Communities of Khatlon Oblast in the Republic of Tajikistan FINAL REPORT Table of Contents Summary Chapter 1 Preface 1.1 Objectives and Scope of the Study .................................................................................. 1 - 1 1.2 Arrangement of Small Hydropower Potential Sites ......................................................... 1 - 2 1.3 Flowchart of the Study Implementation ........................................................................... 1 - 7 Chapter 2 Overview of Energy Situation in Tajikistan 2.1 Economic Activities and Electricity ................................................................................ 2 - 1 2.1.1 Social and Economic situation in Tajikistan ....................................................... 2 - 1 2.1.2 Energy and Electricity ......................................................................................... 2 - 2 2.1.3 Current Situation and Planning for Power Development .................................... 2 - 9 2.2 Natural Condition ............................................................................................................ -

Analysis of Essential Oil from Leaves and Bulbs of Allium Atroviolaceum
Brief Communication and Method report 2020;3(1):e8 Analysis of essential oil from leaves and Bulbs of Allium atroviolaceum a a b c* Parniyan Sebtosheikh , Mahnaz Qomi , Shima Ghadami , Faraz Mojab a. Faculty of Pharmaceutical Chemistry, Pharmaceutical Sciences Branch, Islamic Azad University, Tehran, Iran. b. Faculty of Pharmacy, Pharmaceutical Sciences Branch, Islamic Azad University, Tehran, Iran. c. School of Pharmacy and Pharmaceutical Sciences Research Center, Shahid Beheshti University of Medical Sciences, Tehran, Iran. Article Info: Abstract: Received: September 2020 Introduction: Medicinal plants used in traditional medicine as prevention and treatment Accepted: September 2020 of disease and illness or use in foods, has a long history. Plants belonging to genera Published online: Allium have widely been acquired as food and medicine. In many countries, including September 2020 Iran, a variety of species of the genus Allium such as garlic, onions, leeks, shallots, etc use for food and medicinal uses. Methods and Results: The leaves and bulbs of Allium atroviolaceum, collected from * Corresponding Author: Borujerd (Lorestan Province, Iran) in May 2015 and their essential oils of were obtained Faraz Mojab Email: [email protected] by hydro-distillation. The oils were analyzed by gas chromatography coupled with mass spectrometry (GC/MS) and their chemical composition was identified. The major constituents of A. atroviolaceum leaves oil were dimethyl trisulfide (59.0%), ethyl linolenate (12.4%), phytol (11.4%) and in bulb oil were methyl methyl thiomethyl disulfide (61.3%), dimethyl trisulfide (15.1%) and methyl allyl disulfide (4.3%). The major constituents of both essential oils are sulfur compounds. Conclusion: The results of the present study can help to increase of our information about composition of an edible herb in Iran. -

Nematicidal, Phytotoxic and Brine Shrimp Lethality Activity of Some Allium Species and Their Bioactive Sulfur Compounds
Nematicidal, Phytotoxic and Brine Shrimp Lethality Activity of Some Allium Species and Their Bioactive Sulfur Compounds Dissertation zur Erlangung des Doktorgrades der Naturwissenschaften (Dr. rer. nat.) dem Fachbereich Pharmazie der Philipps-Universität Marburg vorgelegt von Sevda Jivishova aus Baku, Aserbaidschan Marburg/Lahn Jahr 2018 Erstgutachter: Prof. Dr. Michael Keusgen Zweitgutachter: Prof. Dr. Shuming Li Eingereicht am ........................ Tag der ndlichen Prüfung am 21.12.2016 Hochschulkennziffer: 1180 Dedicated to my husband and life partner Emil, our little hearts-children Said and Esma, my beloved parents and my proud brother Pervin, to the supporting parents-in-law and brother-in-law Orkhan. If I have seen further than others, it is by standing upon the shoulders of giants. Isaac Newton TABLE OF CONTENTS TABLE OF CONTENTS ........................................................................................... 1 Acknowledgments .................................................................................................... 5 List of Figures........................................................................................................... 7 List of Tables .......................................................................................................... 10 List of Abbreviations ............................................................................................... 11 Summary ................................................................................................................ 14 -

ISTA List of Stabilized Plant Names 7Th Edition
ISTA List of Stabilized Plant Names th 7 Edition ISTA Nomenclature Committee Chair: Dr. M. Schori Published by All rights reserved. No part of this publication may be The Internation Seed Testing Association (ISTA) reproduced, stored in any retrieval system or transmitted Zürichstr. 50, CH-8303 Bassersdorf, Switzerland in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, without prior ©2020 International Seed Testing Association (ISTA) permission in writing from ISTA. ISBN 978-3-906549-77-4 ISTA List of Stabilized Plant Names 1st Edition 1966 ISTA Nomenclature Committee Chair: Prof P. A. Linehan 2nd Edition 1983 ISTA Nomenclature Committee Chair: Dr. H. Pirson 3rd Edition 1988 ISTA Nomenclature Committee Chair: Dr. W. A. Brandenburg 4th Edition 2001 ISTA Nomenclature Committee Chair: Dr. J. H. Wiersema 5th Edition 2007 ISTA Nomenclature Committee Chair: Dr. J. H. Wiersema 6th Edition 2013 ISTA Nomenclature Committee Chair: Dr. J. H. Wiersema 7th Edition 2019 ISTA Nomenclature Committee Chair: Dr. M. Schori 2 7th Edition ISTA List of Stabilized Plant Names Content Preface .......................................................................................................................................................... 4 Acknowledgements ....................................................................................................................................... 6 Symbols and Abbreviations .......................................................................................................................... -

The Biochemical and Physiological Genesis of Alliin in Garlic
Medicinal and Aromatic Plant Science and Biotechnology ©2007 Global Science Books The Biochemical and Physiological Genesis of Alliin in Garlic M.G. Jones1 • H.A. Collin1 • A. Tregova1 • L. Trueman2 • L. Brown2 • R. Cosstick3 • J. Hughes1 • J. Milne1 • M.C. Wilkinson1 • A.B. Tomsett1 • B. Thomas2* 1 School of Biological Sciences, The Bioscience Building, Crown Street, The University of Liverpool, Liverpool, L69 7ZB, United Kingdom 2 Warwick HRI, University of Warwick, Wellesbourne, Warwick, CV35 9EF, United Kingdom 3 Department of Chemistry, Crown Street, The University of Liverpool, Liverpool, L69 7ZD, United Kingdom Corresponding author : * [email protected] ABSTRACT The health-giving properties of garlic are thought to be primarily derived from the presence and subsequent breakdown of the alk(en)ylcysteine sulphoxide (CSO), alliin and its subsequent breakdown to allicin. Two biosynthetic pathways have been proposed for CSOs, one proceeds from alkylation of glutathione through -glutamyl peptides to yield S-alkyl cysteine sulphoxides while the alternative is direct thioalkylation of serine followed by oxidation to the sulphoxide. Addition of allyl thiol to differentiating garlic tissue cultures resulted in the appearance of detectable levels of both S-allyl cysteine and alliin and also demonstrated that S-allyl-cysteine was oxidised stereospecifically to (+)-alliin by garlic tissue cultures, indicating the presence of a specific oxidase in the cells. Although these reports provide good evidence that S-allyl cysteine can be converted to alliin by garlic tissue cultures, it does not indicate whether "–glutamyl-S- allyl cysteine or S-allyl cysteine is the substrate for oxidation in vivo. Garlic contains several cysteine synthases and at least one has the capacity to synthesise alliin. -

Effect of Extract of Allium Stipitatum on Excisional Wound Healing in Rats Amin Mohammadi-Rika1, Mandana Beigi-Boroujeni2, Asghar Rajabzadeh2, Leila Zarei2,3*
Iran J Vet Surg 2021; 16(1); Serial No: 34; Pages: 5-11 Iranian Veterinary Surgery Association Iranian Journal of Veterinary Surgery Journal homepage: www.ivsajournals.com Original Article Effect of Extract of Allium stipitatum on Excisional Wound Healing in Rats Amin Mohammadi-Rika1, Mandana Beigi-Boroujeni2, Asghar Rajabzadeh2, Leila Zarei2,3* 1 Student Research Committee, Lorestan University of Medical Sciences, Khorramabad, Iran. 2 Department of Anatomical Sciences, Faculty of Medicine, Lorestan University of Medical Sciences, Khorramabad, Iran. 3 Razi Herbal Medicines Research Center, Lorestan University of Medical Sciences, Khorramabad, Iran. ARTICLE INFO ABSTRACT The aim of the present study was to assess the wound-healing activity of extract of Allium Article History: stipitatum. Thirty-six male Wistar rats were used in this study. The rats weighing Received 16 June 2020 approximately 160-180 g and seven weeks of age were randomized into three groups of 12 Revised 20 September 2020 rats each: Control surgery group (Control) including the creation of wounds and no treatment, Accepted 28 September 2020 base formulation groups positive (POS) with the creation of wounds and application of base Online 28 September 2020 formulation ointment, treatment group 1 (T1) with 2 g of powder extract of the plant material in the ointment. A wound was induced by an excisional based wound model in male rats. The Keywords: mature green leaves of Allium stipitatum were collected and authenticated. Extractions of dried leaves were carried out. For wound-healing activity, the extracts were applied topically Herbal extract in the form of ointment and compared to control groups. The healing of the wound was Allium stipitatum assessed based on the wound area, histomorphometry, and hydroxyproline estimation Wound healing studies. -
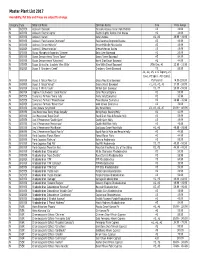
Master Plant List 2017.Xlsx
Master Plant List 2017 Availability, Pot Size and Prices are subject to change. Category Type Botanical Name Common Name Size Price Range N BREVER Azalea X 'Cascade' Cascade Azalea (Glenn Dale Hybrid) #3 49.99 N BREVER Azalea X 'Electric Lights' Electric Lights Double Pink Azalea #2 44.99 N BREVER Azalea X 'Karen' Karen Azalea #2, #3 39.99 - 49.99 N BREVER Azalea X 'Poukhanense Improved' Poukhanense Improved Azalea #3 49.99 N BREVER Azalea X 'Renee Michelle' Renee Michelle Pink Azalea #3 49.99 N BREVER Azalea X 'Stewartstonian' Stewartstonian Azalea #3 49.99 N BREVER Buxus Microphylla Japonica "Gregem' Baby Gem Boxwood #2 29.99 N BREVER Buxus Sempervirens 'Green Tower' Green Tower Boxwood #5 64.99 N BREVER Buxus Sempervirens 'Katerberg' North Star Dwarf Boxwood #2 44.99 N BREVER Buxus Sinica Var. Insularis 'Wee Willie' Wee Willie Dwarf Boxwood Little One, #1 13.99 - 21.99 N BREVER Buxus X 'Cranberry Creek' Cranberry Creek Boxwood #3 89.99 #1, #2, #5, #15 Topiary, #5 Cone, #5 Spiral, #10 Spiral, N BREVER Buxus X 'Green Mountain' Green Mountain Boxwood #5 Pyramid 14.99-299.99 N BREVER Buxus X 'Green Velvet' Green Velvet Boxwood #1, #2, #3, #5 17.99 - 59.99 N BREVER Buxus X 'Winter Gem' Winter Gem Boxwood #5, #7 59.99 - 99.99 N BREVER Daphne X Burkwoodii 'Carol Mackie' Carol Mackie Daphne #2 59.99 N BREVER Euonymus Fortunei 'Ivory Jade' Ivory Jade Euonymus #2 35.99 N BREVER Euonymus Fortunei 'Moonshadow' Moonshadow Euonymus #2 29.99 - 35.99 N BREVER Euonymus Fortunei 'Rosemrtwo' Gold Splash Euonymus #2 39.99 N BREVER Ilex Crenata 'Sky Pencil' -

Alliinase from Ensifer Adhaerens and Its Use for Generation of Fungicidal
Yutani et al. AMB Express 2011, 1:2 http://www.amb-express.com/content/1/1/2 ORIGINALARTICLE Open Access Alliinase from Ensifer adhaerens and Its Use for Generation of Fungicidal Activity Masahiro Yutani1, Hiroko Taniguchi1, Hasibagan Borjihan1, Akira Ogita1,2, Ken-ichi Fujita1, Toshio Tanaka1* Abstract AbacteriumEnsifer adhaerens FERM P-19486 with the ability of alliinase production was isolated from a soil sample. The enzyme was purified for characterization of its general properties and evaluation of its application in on-site production of allicin-dependent fungicidal activity. The bacterial alliinase was purified 300-fold from a cell-free extract, giving rise to a homogenous protein band on polyacrylamide gel electrophoresis. The bacterial alliinase (96 kDa) consisted of two identical subunits (48 kDa), and was most active at 60°C and at pH 8.0. The enzyme stoichiometrically converted (-)-alliin ((-)-S-allyl-L-cysteine sulfoxide) to form allicin, pyruvic acid, and ammonia more selectively than (+)-alliin, a naturally occurring substrate for plant alliinase ever known. The C-S lyase activity was also detected with this bacterial enzyme when S-alkyl-L-cysteine was used as a substrate, though such a lyase activity is absolutely absent in alliinase of plant origin. The enzyme generated a fungicidal activity against Saccharomyces cerevisiae in a time- and a dose-dependent fashion using alliin as a stable precursor. Alliinase of Ensifer adhaerens FERM P-19486 is the enzyme with a novel type of substrate specificity, and thus considered to be beneficial when used in combination with garlic enzyme with respect to absolute conversion of (±)-alliin to allicin. -

Survey of Wild Food Plants for Human Consumption in Geçitli (Hakkari, Turkey)
Indian Journal of Traditional Knowledge Vol. 14(2), April 2015, pp. 183-190 Survey of wild food plants for human consumption in Geçitli (Hakkari, Turkey) İdris Kaval1, Lütfi Behçet2 & Uğur Çakilcioğlu3* 1Yuzuncu Yıl University, Department of Biology, Van 65000, Turkey; 2Bingöl University, Department of Biology, Bingöl 12000, Turkey; 3Tunceli University, Pertek Sakine Genç Vocational School, Pertek, Tunceli 62500, Turkey E-mails: [email protected]; [email protected]; [email protected] Received 15 July 2014, revised 22 January 2015 This study aims to record accumulation of knowledge on plants which are used as food by native people of Geçitli (Hakkari, Turkey) that has a rich culture and a very natural environment. In addition, the medical uses of these plants were compiled from the literature. Study area was located on the East of Anatolian diagonal, in the Eastern Anatolia region. Field study was carried out over a period of approximately two years (2008-2010). During this period, 84 vascular plant taxa were collected. The plants were pressed in the field and prepared for identification. A total of 84 food plants belonging to 30 families were identified in the region. In the study being conducted, use of wild plants as food points out interest of people in Geçitli in wild plants. The fact that a large proportion of edible plants are also being used for medicinal purposes indicates that the use of wild plants has a high potential in the region. The present study shows that further ethnobotanical investigations are worthy to be carried out in Turkey, where most of knowledge on popular food plants are still to discover.