Genomics of a Complete Butterfly Continent
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ancient Roaches Further Exemplify 'No Land Return' in Aquatic Insects
Gondwana Research 68 (2019) 22–33 Contents lists available at ScienceDirect Gondwana Research journal homepage: www.elsevier.com/locate/gr Ancient roaches further exemplify ‘no land return’ in aquatic insects Peter Vršanský a,b,c,d,1, Hemen Sendi e,⁎,1, Danil Aristov d,f,1, Günter Bechly g,PatrickMüllerh, Sieghard Ellenberger i, Dany Azar j,k, Kyoichiro Ueda l, Peter Barna c,ThierryGarciam a Institute of Zoology, Slovak Academy of Sciences, Dúbravská cesta 9, 845 06 Bratislava, Slovakia b Slovak Academy of Sciences, Institute of Physics, Research Center for Quantum Information, Dúbravská cesta 9, Bratislava 84511, Slovakia c Earth Science Institute, Slovak Academy of Sciences, Dúbravská cesta 9, P.O. BOX 106, 840 05 Bratislava, Slovakia d Paleontological Institute, Russian Academy of Sciences, Profsoyuznaya 123, 117868 Moscow, Russia e Faculty of Natural Sciences, Comenius University, Ilkovičova 6, Bratislava 84215, Slovakia f Cherepovets State University, Cherepovets 162600, Russia g Staatliches Museum für Naturkunde Stuttgart, Rosenstein 1, D-70191 Stuttgart, Germany h Friedhofstraße 9, 66894 Käshofen, Germany i Bodelschwinghstraße 13, 34119 Kassel, Germany j State Key Laboratory of Palaeobiology and Stratigraphy, Nanjing Institute of Geology and Palaeontology, Chinese Academy of Sciences, Nanjing 210008, PR China k Lebanese University, Faculty of Science II, Fanar, Natural Sciences Department, PO Box 26110217, Fanar - Matn, Lebanon l Kitakyushu Museum, Japan m River Bigal Conservation Project, Avenida Rafael Andrade y clotario Vargas, 220450 Loreto, Orellana, Ecuador article info abstract Article history: Among insects, 236 families in 18 of 44 orders independently invaded water. We report living amphibiotic cock- Received 13 July 2018 roaches from tropical streams of UNESCO BR Sumaco, Ecuador. -

January 1990
TORREYANA Published for Members of the Torrey Pines Docent Society and the Torrey Pines Association No. 172 January 1990 Next Docent Society Meeting SATURDAY, JANUA..t<¥ 20, 9: 00 A.M. AT THE VISI'IDR CENI'ER David F. Marriott will speak on "Butterflies of the TPSR Area" at the January meeting (alsc> see his article in this nonth 1 s Torreyana). He will show specimens of all species, ·as ...e11 as slides, photos, videos, and rredia coverage. Dr. Marriott is currently teaching a course through UCSD Extension on the butterflies of San Diego County, with an emphasis on rbnarch migration. He has been studying the butterfly species of the TPSR area during the past five years, and has studied and collected nearly all the ~ : sp:cies from San Diego County. For information requested for the business part of the meeting, see the Docent President 1 s notes below . ...~·~ .· Docent President's Notes by Michael K. Fox What a great_Christrnas party! (pictures~ p. 2) On behalf of the docents and directors of t.~ Torrey Pines Docent Society, I wish to extend cor71plirnents and thanks to the class of 1 89 for a job well done. Thanks to each and every one of :you! Since the new year is UPJrl us, it is tirre to do a little retrospective analysis ··-you know, hin£1~~ight. For the January rreeting, the Board of Directors v..ould like to have suggestJ:'6fi~ from the general rrembership for nolT'inations for "Docent of the Year." Please .. either :r;ail your choice to rre or bring it on a 3x5 card to hand in at the rreeting. -

Panicledleaf Ticktrefoil (Desmodium Paniculatum) Plant Fact Sheet
Plant Fact Sheet Hairstreak (Strymon melinus) eat the flowers and PANICLEDLEAF developing seedpods. Other insect feeders include many kinds of beetles, and some species of thrips, aphids, moth TICKTREFOIL caterpillars, and stinkbugs. The seeds are eaten by some upland gamebirds (Bobwhite Quail, Wild Turkey) and Desmodium paniculatum (L.) DC. small rodents (White-Footed Mouse, Deer Mouse), while Plant Symbol = DEPA6 the foliage is readily eaten by White-Tailed Deer and other hoofed mammalian herbivores. The Cottontail Contributed by: USDA, NRCS, Norman A. Berg National Rabbit also consumes the foliage. Plant Materials Center, Beltsville, MD Status Please consult the PLANTS Web site and your State Department of Natural Resources for this plant’s current status (e.g. threatened or endangered species, state noxious status, and wetland indicator values). Description and Adaptation Panicledleaf Ticktrefoil is a native, perennial, wildflower that grows up to 3 feet tall. The genus Desmodium: originates from Greek meaning "long branch or chain," probably from the shape and attachment of the seedpods. The central stem is green with clover-like, oblong, multiple green leaflets proceeding singly up the stem. The showy purple flowers appear in late summer and grow arranged on a stem maturing from the bottom upwards. In early fall, the flowers produce leguminous seed pods approximately ⅛ inch long. Panicledleaf Photo by Rick Mark [email protected] image used with permission Ticktrefoil plants have a single crown. This wildflower is a pioneer species that prefers some disturbance from Alternate Names wildfires, selective logging, and others causes. The sticky Panicled Tick Trefoil seedpods cling to the fur of animals and the clothing of Uses humans and are carried to new locations. -

Biodiversity Work Group Report: Appendices
Biodiversity Work Group Report: Appendices A: Initial List of Important Sites..................................................................................................... 2 B: An Annotated List of the Mammals of Albemarle County........................................................ 5 C: Birds ......................................................................................................................................... 18 An Annotated List of the Birds of Albemarle County.............................................................. 18 Bird Species Status Tables and Charts...................................................................................... 28 Species of Concern in Albemarle County............................................................................ 28 Trends in Observations of Species of Concern..................................................................... 30 D. Fish of Albemarle County........................................................................................................ 37 E. An Annotated Checklist of the Amphibians of Albemarle County.......................................... 41 F. An Annotated Checklist of the Reptiles of Albemarle County, Virginia................................. 45 G. Invertebrate Lists...................................................................................................................... 51 H. Flora of Albemarle County ...................................................................................................... 69 I. Rare -

Self-Repair and Self-Cleaning of the Lepidopteran Proboscis
Clemson University TigerPrints All Dissertations Dissertations 8-2019 Self-Repair and Self-Cleaning of the Lepidopteran Proboscis Suellen Floyd Pometto Clemson University, [email protected] Follow this and additional works at: https://tigerprints.clemson.edu/all_dissertations Recommended Citation Pometto, Suellen Floyd, "Self-Repair and Self-Cleaning of the Lepidopteran Proboscis" (2019). All Dissertations. 2452. https://tigerprints.clemson.edu/all_dissertations/2452 This Dissertation is brought to you for free and open access by the Dissertations at TigerPrints. It has been accepted for inclusion in All Dissertations by an authorized administrator of TigerPrints. For more information, please contact [email protected]. SELF-REPAIR AND SELF-CLEANING OF THE LEPIDOPTERAN PROBOSCIS A Dissertation Presented to the Graduate School of Clemson University In Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy ENTOMOLOGY by Suellen Floyd Pometto August 2019 Accepted by: Dr. Peter H. Adler, Major Advisor and Committee Co-Chair Dr. Eric Benson, Committee Co-Chair Dr. Richard Blob Dr. Patrick Gerard i ABSTRACT The proboscis of butterflies and moths is a key innovation contributing to the high diversity of the order Lepidoptera. In addition to taking nectar from angiosperm sources, many species take up fluids from overripe or sound fruit, plant sap, animal dung, and moist soil. The proboscis is assembled after eclosion of the adult from the pupa by linking together two elongate galeae to form one tube with a single food canal. How do lepidopterans maintain the integrity and function of the proboscis while foraging from various substrates? The research questions included whether lepidopteran species are capable of total self- repair, how widespread the capability of self-repair is within the order, and whether the repaired proboscis is functional. -

Predaceous Ground Beetles Caterpillar Hunters and Bombardier
E-185 5-03 PredaceousPredaceous GroundGround BeetBeetlesles Caterpillar Hunters and Bombardier Beetles Rick Minzenmayer, Extension Agent-IPM Chris Sansone, Extension Entomologist Texas Cooperative Extension redaceous ground beetles can be a nui- genus Calosoma, a brightly colored ground sance when numerous. They are beetle. Some species are called “bombardier PPattracted to lights and can sometimes beetles” because they emit what appears to be be found by the hundreds around lights in the smoke from the rear of the abdomen. The morning. The large numbers can also be a “smoke” is actually a glandular fluid that problem because the beetles defend them- vaporizes when it hits air; the fluid can irri- selves by emitting an odor. tate the skin. The ground beetles also emit an Ground beetles are part of the order odor to stop their enemies, including people. Coleoptera. This is the largest order of insects with over a quarter of a million species described throughout the world — about 30,000 species in the United States. Most beetles have two pairs of wings (elytra). The front pair is usually thickened and hard and meet in a straight line down the back when the wings are at rest. The back pair are mem- branous and folded beneath the front pair. All beetles have chewing mouthparts and under- go complete metamorphosis (egg, larva, pupa and adult). Predaceous ground beetles belong to the fami- ly Carabidae. This is the second largest family Caterpillar hunter, Calasoma scrutator (Fabricius) (Coleoptera: of beetles, with more than 2,500 species in Carabidae). North America. Most members of this family are considered beneficial, feeding on other insects in both the larval and adult stages. -

Insects Parasitoids: Natural Enemies of Helicoverpa
Queensland the Smart State insects Parasitoids: Natural enemies of helicoverpa Introduction Helicoverpa caterpillars (often called heliothis) are serious pests of many crops in Australia. A range of parasitoid and predatory insects attack helicoverpa. Identifying and conserving these beneficial insects is fundamental to implementing pest management with a reduced reliance on chemical insecticides. This brochure describes the most important parasitoids of helicoverpa in Australian broadacre crops. Parasitoids versus parasites: What’s the difference? Parasitoids kill their hosts; parasites (such Figure 1. Netelia producta is one of the as lice and fleas) do not. All the insects most commonly encountered parasitoids in this brochure are parasitoids. Despite of helicoverpa. Females lay their eggs onto this difference, the terms parasitoid and caterpillars, and the hatching wasp larva parasite are often used interchangeably, if feeds on its host, eventually killing it. inaccurately. Parasitoids such as Netelia can be important biological control agents of helicoverpa in crops. (Photo: K. Power) All comments about parasitoid abundance in this publication are based on field observations in southern Queensland farming systems. These patterns may not occur in all parts of Australia. About parasitoids What is a parasitoid? How do parasitoids find their A parasitoid is an insect that kills (parasitises) hosts? its host — usually another insect — in Many adult parasitoids find their host by order to complete its lifecycle. In Australia, smell. They can detect the direct odour of helicoverpa are parasitised by many species the host itself, or odours associated with host of wasps and flies. All helicoverpa immature activity, such as plant damage or caterpillar stages are parasitised (that is, egg, caterpillar frass (dung). -

“Sockets” Or Are Articulated, As Are the Primary D Or SD GIELIS, C
VOLUME 60, NUMBER 2 97 “sockets” or are articulated, as are the primary D or SD GIELIS, C. 2000. Division of the Pterophoridae into tribes (Lepi- setae. doptera). Quadrifina 3: 57-60. GIELIS, C. 2003. Pterophoridae & Alucitoidea - In: World Catalogue While tribal placement of Cosmoclostis in of Insects 4: 1-198. Pterophorini can be supported, additional species need HEINRICH, C. 1916. On the taxonomic value of some larval characters to be examined, to ascertain larval and pupal characters in the Lepidoptera. Proc. Entomol. Soc. Wash. 18: 154-164. HAO, S.-L., LI, H.-H. , & WU, C.-S. 2004. First record of the genus entirely unique to the genus Cosmoclostis. Continuing Cosmoclostis Meyrick from China, with descriptions of two new studies of larval and pupal structure will provide species (Lepidoptera, Pterophoridae). Acta Zoo. Sinica 29(1): additional insight into the relationships between and 142-146. MATTHEWS, D.L. & LOTT, T.A. 2005. Larval Hostplants of the within tribes and subfamilies of this group. Pterophoridae (Lepidoptera: Pterophoroidea). Mem. Amer. En- tomol. Inst. 76: 1-324. We thank Marianne Horak, Australian National Insect Collection, MEYRICK, E. 1886. On the classification of the Pterophoridae. Trans. for arranging the loan of the described specimens. George Mathew, Entom. Soc. Lond. 1886: 1-21. Kerala Forest Institute, India, provided comparative larval material of MOSHER, E. 1916. A classification of the Lepidoptera based on char- Cosmoclostis leucomochla. We also thank Terry A. Lott, J. Howard acters of the pupa. Bull. Ill. State Lab. Nat. Hist. 12(2): 18-153, Frank, and Donald W. Hall for reviewing the manuscript. -

Butterflies of the Wesleyan Campus
BUTTERFLIES OF THE WESLEYAN CAMPUS SWALLOWTAILS Hairstreaks (Subfamily - Theclinae) (Family PAPILIONIDAE) Great Purple Hairstreak - Atlides halesus Coral Hairstreak - Satyrium titus True Swallowtails Banded Hairstreak - Satyrium calanus (Subfamily - Papilioninae) Striped Hairstreak - Satyrium liparops Pipevine Swallowtail - Battus philenor Henry’s Elfin - Callophrys henrici Zebra Swallowtail - Eurytides marcellus Eastern Pine Elfin - Callophrys niphon Black Swallowtail - Papilio polyxenes Juniper Hairstreak - Callophrys gryneus Giant Swallowtail - Papilio cresphontes White M Hairstreak - Parrhasius m-album Eastern Tiger Swallowtail - Papilio glaucus Gray Hairstreak - Strymon melinus Spicebush Swallowtail - Papilio troilus Red-banded Hairstreak - Calycopis cecrops Palamedes Swallowtail - Papilio palamedes Blues (Subfamily - Polommatinae) Ceraunus Blue - Hemiargus ceraunus Eastern-Tailed Blue - Everes comyntas WHITES AND SULPHURS Spring Azure - Celastrina ladon (Family PIERIDAE) Whites (Subfamily - Pierinae) BRUSHFOOTS Cabbage White - Pieris rapae (Family NYMPHALIDAE) Falcate Orangetip - Anthocharis midea Snouts (Subfamily - Libytheinae) American Snout - Libytheana carinenta Sulphurs and Yellows (Subfamily - Coliadinae) Clouded Sulphur - Colias philodice Heliconians and Fritillaries Orange Sulphur - Colias eurytheme (Subfamily - Heliconiinae) Southern Dogface - Colias cesonia Gulf Fritillary - Agraulis vanillae Cloudless Sulphur - Phoebis sennae Zebra Heliconian - Heliconius charithonia Barred Yellow - Eurema daira Variegated Fritillary -

Genomic Analysis of the Tribe Emesidini (Lepidoptera: Riodinidae)
Zootaxa 4668 (4): 475–488 ISSN 1175-5326 (print edition) https://www.mapress.com/j/zt/ Article ZOOTAXA Copyright © 2019 Magnolia Press ISSN 1175-5334 (online edition) https://doi.org/10.11646/zootaxa.4668.4.2 http://zoobank.org/urn:lsid:zoobank.org:pub:211AFB6A-8C0A-4AB2-8CF6-981E12C24934 Genomic analysis of the tribe Emesidini (Lepidoptera: Riodinidae) JING ZHANG1, JINHUI SHEN1, QIAN CONG1,2 & NICK V. GRISHIN1,3 1Departments of Biophysics and Biochemistry, University of Texas Southwestern Medical Center, and 3Howard Hughes Medical Insti- tute, 5323 Harry Hines Blvd, Dallas, TX, USA 75390-9050; [email protected] 2present address: Institute for Protein Design and Department of Biochemistry, University of Washington, 1959 NE Pacific Street, HSB J-405, Seattle, WA, USA 98195; [email protected] Abstract We obtained and phylogenetically analyzed whole genome shotgun sequences of nearly all species from the tribe Emesidini Seraphim, Freitas & Kaminski, 2018 (Riodinidae) and representatives from other Riodinidae tribes. We see that the recently proposed genera Neoapodemia Trujano, 2018 and Plesioarida Trujano & García, 2018 are closely allied with Apodemia C. & R. Felder, [1865] and are better viewed as its subgenera, new status. Overall, Emesis Fabricius, 1807 and Apodemia (even after inclusion of the two subgenera) are so phylogenetically close that several species have been previously swapped between these two genera. New combinations are: Apodemia (Neoapodemia) zela (Butler, 1870), Apodemia (Neoapodemia) ares (Edwards, 1882), and Apodemia (Neoapodemia) arnacis (Stichel, 1928) (not Emesis); and Emesis phyciodoides (Barnes & Benjamin, 1924) (not Apodemia), assigned to each genus by their monophyly in genomic trees with the type species (TS) of the genus. -
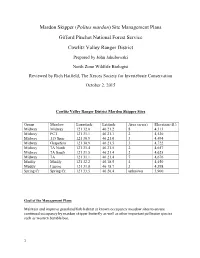
Mardon Skipper Site Management Plans
Mardon Skipper (Polites mardon) Site Management Plans Gifford Pinchot National Forest Service Cowlitz Valley Ranger District Prepared by John Jakubowski North Zone Wildlife Biologist Reviewed by Rich Hatfield, The Xerces Society for Invertebrate Conservation October 2, 2015 Cowlitz Valley Ranger District Mardon Skipper Sites Group Meadow Longitude Latitude Area (acres) Elevation (ft.) Midway Midway 121 32.0 46 21.2 8 4,313 Midway PCT 121 31.1 46 21.1 2 4,530 Midway 115 Spur 121 30.9 46 21.0 3 4,494 Midway Grapefern 121 30.9 46 21.5 3 4,722 Midway 7A North 121 31.4 46 21.5 2 4,657 Midway 7A South 121 31.5 46 21.4 2 4,625 Midway 7A 121 31.1 46 21.4 7 4,676 Muddy Muddy 121 32.2 46.18.5 4 4,450 Muddy Lupine 121 31.8 46 18.7 3 4,398 Spring Cr Spring Cr. 121 33.5 46 20.4 unknown 3,900 Goal of the Management Plans Maintain and improve grassland/forb habitat at known occupancy meadow sites to ensure continued occupancy by mardon skipper butterfly as well as other important pollinator species such as western bumble bee. 1 Introduction On the Gifford Pinchot National Forest (GPNF), mardon skippers were first detected on the Mt. Adams Ranger District (MTA) in 2000 and on Cowlitz Valley Ranger District (CVRD) in 2002. Mardon skippers are known to inhabit ten, upland dry grassy meadows on the CVRD. Portions of the meadows are mesic and are unsuitable mardon skipper habitat. -

A New Sandy-Desert Subspecies of Megathymus Coloradensis (Megathymidae) from Extreme Northern Arizona
BULLETIN OF THE ALLYN MUSEUM Published by THE ALLYN MUSEUM OF ENTOMOLOGY Sarasota, Florida Number -- 17 12 APRIL 1974 A NEW SANDY-DESERT SUBSPECIES OF MEGATHYMUS COLORADENSIS (MEGATHYMIDAE) FROM EXTREME NORTHERN ARIZONA Ronald S.' and Dale Wielgus 3434 West Augusta Avenue, Phoenix, Arizona 85021 Recent descriptions in the genus Cercyonis (Satyridae) by Emmel & Emmel (1969; 1971) and Emmel & Mattoon (1972) have shown the existence of distinctive whitish races adapted to arid alkaline flats of the western deserts of the United States. Such environmental adaptation appears to extend to a population of Megathymus coloradensis Riley inhabiting a sandy-desert area of extreme northern Arizona. The discovery of the subspecies described herein was a result of our initial investigations into the biology of Megathymus streckeri (Skinner). The purpose of this paper is to describe the new population and to note its life history. This study considered the following subspecies: Megathymus coloradensis navajo Skinner, Megathymus coloradensis arizonae Tinkham, Megathymus coloradensis reubeni Stallings, Turner & Stallings, Megathymus coloradensis coloradensis Riley and Megathymus coloradensis browni Stallings & Turner. Megathymus coloradensis albasuffusa Ronald S. and Dale Wielgus, new subspecies. Female. Head: vertex grey, Palpus: white, with some black-tipped scales. Antenna: shaft white distad, ringed with black basad; club black. Thorax: grey dorsally with long greenish-brown hairs caudad, blackish ventrally. Abdomen: black dorsally, grey ventrally.