Arxiv:2010.04222V1 [Cond-Mat.Mtrl-Sci] 8 Oct 2020
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Semantic Skin: from Flat Textual Content to Interconnected Repositories Of
Semantic Skin: from flat textual content to interconnected repositories of semantic data. Claudio Baldassarre ABSTRACT front-end web application. This application offers a faceted One approach to re-balancing the Digital Divide tends to view of the underlying \news-KB". The current blog site favor the production of informative content in flat formats, appearance is merely a stylistic choice, while a running in- which are easy to distribute and consume. At the same time stance is always backed by a SPARQL endpoint over the this approach forbids to deliver the core knowledge perti- \news-KB". The facets are typically rendered as menu ele- nent within the content; i.e. it increases the Knowledge ments5: some menus facet the entire \news-KB" (e.g., news Divide. In some international organizations1, informative Topics, or Provenance); while other menus facet only the content distribution to groups in Latin America happens by content currently visible to the users. The faceting mech- manually collecting text-based content, then disseminating anism is also applied tothe \news archive" as a time-based it via standard mailing lists, or databases copies sent out facet of the repository content. All the facets are popu- regularly. Our demo showcases the use of Semantic Skin lated with SPARQL queries over the \news-model" instances a technology that after semantifying the content submit- in the \news-KB". Each news item is then presented with ted in flat formats, provides access to the information via a its summary, title, publication date, and provenance (e.g., knowledge layer, which is, however, transparent to the end permalink). -

Where Is the Semantic Web? – an Overview of the Use of Embeddable Semantics in Austria
Where Is The Semantic Web? – An Overview of the Use of Embeddable Semantics in Austria Wilhelm Loibl Institute for Service Marketing and Tourism Vienna University of Economics and Business, Austria [email protected] Abstract Improving the results of search engines and enabling new online applications are two of the main aims of the Semantic Web. For a machine to be able to read and interpret semantic information, this content has to be offered online first. With several technologies available the question arises which one to use. Those who want to build the software necessary to interpret the offered data have to know what information is available and in which format. In order to answer these questions, the author analysed the business websites of different Austrian industry sectors as to what semantic information is embedded. Preliminary results show that, although overall usage numbers are still small, certain differences between individual sectors exist. Keywords: semantic web, RDFa, microformats, Austria, industry sectors 1 Introduction As tourism is a very information-intense industry (Werthner & Klein, 1999), especially novel users resort to well-known generic search engines like Google to find travel related information (Mitsche, 2005). Often, these machines do not provide satisfactory search results as their algorithms match a user’s query against the (weighted) terms found in online documents (Berry and Browne, 1999). One solution to this problem lies in “Semantic Searches” (Maedche & Staab, 2002). In order for them to work, web resources must first be annotated with additional metadata describing the content (Davies, Studer & Warren., 2006). Therefore, anyone who wants to provide data online must decide on which technology to use. -

Appendix a the Ten Commandments for Websites
Appendix A The Ten Commandments for Websites Welcome to the appendixes! At this stage in your learning, you should have all the basic skills you require to build a high-quality website with insightful consideration given to aspects such as accessibility, search engine optimization, usability, and all the other concepts that web designers and developers think about on a daily basis. Hopefully with all the different elements covered in this book, you now have a solid understanding as to what goes into building a website (much more than code!). The main thing you should take from this book is that you don’t need to be an expert at everything but ensuring that you take the time to notice what’s out there and deciding what will best help your site are among the most important elements of the process. As you leave this book and go on to updating your website over time and perhaps learning new skills, always remember to be brave, take risks (through trial and error), and never feel that things are getting too hard. If you choose to learn skills that were only briefly mentioned in this book, like scripting, or to get involved in using content management systems and web software, go at a pace that you feel comfortable with. With that in mind, let’s go over the 10 most important messages I would personally recommend. After that, I’ll give you some useful resources like important websites for people learning to create for the Internet and handy software. Advice is something many professional designers and developers give out in spades after learning some harsh lessons from what their own bitter experiences. -
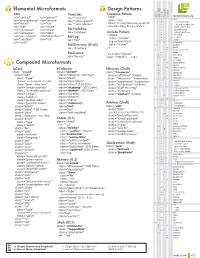
Microformats Cheat Sheet
Elemental Microformats Design Patterns tom eview Datetime Pattern esume XFN VoteLinks microformats.org hR hR hCard hCalendar rel="contact" rel="parent" rev="vote-for" <abbr hA rel="acquaintance" rel="spouse" class="foo" • adr rev="vote-against" + country-name title="YYYY-MM-DDTHH:MM:SS+ZZ:ZZ" rel="friend" rel="kin" rev="vote-abstain" • extended-address rel="met" rel="muse" >Human Date Time</abbr> + post-office-box rel="co-worker" rel="crush" Rel-Nofollow + postal-code rel="colleague" rel="date" rel="nofollow" Include Pattern • street-address + locality rel="co-resident" rel="sweetheart" <object Rel-Tag class="include" + region rel="neighbor" rel="me" rel="tag" • type rel="child" type="text/html" • affiliation Rel-Directory (Draft) data="#idref" ¤ author rel="directory" /> + best × + bookmark (rel) Rel-License + bday <a class="include" • • category rel="license" href="#idref">...</a> + + class × contact Compound Microformats + description + dtend + dtreviewed hCard hCalendar hResume (Draft) × dtstart class="vcard" class="vevent" class="hresume" × dtstamp class="adr" class="category" rel="tag" class="affiliation" (hcard) duration class="type" class="class" class="education" (hcalendar) • education [work|home|pref|postal|dom|intl] class="description" • email class="experience" (hcalendar) • type class="post-office-box" class="dtend" (ISO Date) class="publication" (citation) • value class="street-address" class="dtstamp" (ISO Date) class="skill" rel="tag" × entry-content class="extended-address" class="dtstart" (ISO Date) class="summary" • entry-summary -

Microformats: Empowering Your Markup for Web 2.0
Microformats: Empowering Your Markup for Web 2.0 John Allsopp Microformats: Empowering Your Markup for Web 2.0 Copyright © 2007 by John Allsopp All rights reserved. No part of this work may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopying, recording, or by any information storage or retrieval system, without the prior written permission of the copyright owner and the publisher. ISBN-13 (pbk): 978-1-59059814-6 ISBN-10 (pbk): 1-59059-814-8 Printed and bound in the United States of America 9 8 7 6 5 4 3 2 1 Trademarked names may appear in this book. Rather than use a trademark symbol with every occurrence of a trademarked name, we use the names only in an editorial fashion and to the benefit of the trademark owner, with no intention of infringement of the trademark. Distributed to the book trade worldwide by Springer-Verlag New York, Inc., 233 Spring Street, 6th Floor, New York, NY 10013. Phone 1-800-SPRINGER, fax 201-348-4505, e-mail [email protected],or visit www.springeronline.com. For information on translations, please contact Apress directly at 2560 Ninth Street, Suite 219, Berkeley, CA 94710. Phone 510-549-5930, fax 510-549-5939, e-mail [email protected], or visit www.apress.com. The information in this book is distributed on an “as is” basis, without warranty. Although every precaution has been taken in the preparation of this work, neither the author(s) nor Apress shall have any liability to any person or entity with respect to any loss or damage caused or alleged to be caused directly or indirectly by the information contained in this work. -

Standardization's All Very Well, but What About the Exabytes of Existing
Standardization’s all very well, but what about the Exabytes of Existing Content and Learner Contributions? Felix Mödritscher 1), Victor Manuel García-Barrios 2) 1) Institute for Information Systems and New Media, Vienna University of Economics and Business Administration, Augasse 2-6, 1090 Vienna, Austria [email protected] 2) Institute for Information Systems and Computer Media, Graz University of Technology, Inffeldgasse 16c, 8010 Graz, Austria [email protected] 1. Problem Definition Standardization in the field of technology-enhanced learning focuses on structuring and aggregating assets to interoperable educational entities, like a course package. However, available standards and specifications in this area do not include an approach for addressing semantics embedded in existing content. This consideration might be useful for user-centered concepts, like learners tagging or commenting existing material, as well as for automated mechanism, like extracting relations or other meta-information automatically from the resources. In the upcoming section we indicate application areas and explain why available standards and specifications do not support these scenarios. Thereafter, we present a XML- based description language for semantics embedded in web-based content, which might be a solution for these use cases. Finally, we summarize and discuss some experiences on in- content semantics from former projects, particularly AdeLE (Adaptive e-Learning with Eye- tracking, http://adele.fh-joanneum.at) and iCamp (http://icamp.eu), and give an outlook on future work. 2. Application Areas and Shortcomings of Standards In 2004 we came in the situation that we had to cope with semantic enrichment of existing learning content, precisely to enable facilitators to tag web-based resources. -

The Global Rock-Art Database
The Global Rock-Art Database: Centralizing Heritage Data Collections using a Collaborative, Information Structure and Data Visualization Approach in an Open Source Application Author Haubt, Robert Alexander Published 2016 Thesis Type Thesis (PhD Doctorate) School School of Humanities, Languages and Social Science DOI https://doi.org/10.25904/1912/3082 Copyright Statement The author owns the copyright in this thesis, unless stated otherwise. Downloaded from http://hdl.handle.net/10072/367148 Griffith Research Online https://research-repository.griffith.edu.au The Global Rock-Art Database Centralizing Heritage Data Collections using a Collaborative, Information Structure and Data Visualization Approach in an Open Source Application Mr. Robert Alexander Haubt BA (Hons), MA School of Humanities, Languages and Social Sciences Griffith University Submitted in fulfillment of the requirements of the degree of Doctor of Philosophy May 2016 Statement of Originality This work has not previously been submitted for a degree or diploma in any university. To the best of my knowledge and belief, the thesis contains no material previously published or written by another person except where due reference is made in the thesis itself. _____________________________ Robert A. Haubt I Acknowledgments Acknowledgments Firstly, I would like to express my sincere gratitude to my supervisor Prof. P.S.C. Taçon for the support of my PhD research during the last four years. His patience, motivation and knowledge helped guide me through my research and I could not have imagined a better mentor for my PhD studies. Besides my supervisor, I would like to thank Dr. Duncan Wright, Dr. Tim Humphrey and Dr. Jason Nelson for their guidance and support as my associate supervisors. -

Web Standards.Pdf
BOOKS FOR PROFESSIONALS BY PROFESSIONALS® Sikos, Ph.D. RELATED Web Standards Web Standards: Mastering HTML5, CSS3, and XML gives you a deep understand- ing of how web standards can be applied to improve your website. You will also find solutions to some of the most common website problems. You will learn how to create fully standards-compliant websites and provide search engine-optimized Web documents with faster download times, accurate rendering, lower development costs, and easy maintenance. Web Standards: Mastering HTML5, CSS3, and XML describes how you can make the most of web standards, through technology discussions as well as practical sam- ple code. As a web developer, you’ll have seen problems with inconsistent appearance and behavior of the same site in different browsers. Web standards can and should be used to completely eliminate these problems. With Web Standards, you’ll learn how to: • Hand code valid markup, styles, and news feeds • Provide meaningful semantics and machine-readable metadata • Restrict markup to semantics and provide reliable layout • Achieve full standards compliance Web standardization is not a sacrifice! By using this book, we can create and maintain a better, well-formed Web for everyone. CSS3, and XML CSS3, Mastering HTML5, US $49.99 Shelve in Web Development/General User level: Intermediate–Advanced SOURCE CODE ONLINE www.apress.com www.it-ebooks.info For your convenience Apress has placed some of the front matter material after the index. Please use the Bookmarks and Contents at a Glance links to access them. www.it-ebooks.info Contents at a Glance About the Author................................................................................................ -

Cinii: Bringing Linked Data to Japan's Largest Scholarly Search Engine
Proc. Int’l Conf. on Dublin Core and Metadata Applications 2009 CiNii: Bringing Linked Data to Japan's Largest Scholarly Search Engine Ikki Ohmukai Hideaki Takeda National Institute of Informatics, National Institute of Informatics, Japan Japan [email protected] [email protected] Keywords: metadata; linked data; academic information services; OpenSearch; RDF. Abstract National Institute of Informatics operates "CiNii" (http://ci.nii.ac.jp/), the largest scholarly search engine in Japan. CiNii is a database of journals and proceedings. It stores full text and bibliography of over 3 million articles. In cooperation with National Diet Library, electronic journal publishers and institutional repositories, CiNii also collects bibliographic metadata of 20 million articles. The number of page views was over 10 million in December 2008 and still increasing. Figure1 shows screenshots of CiNii. CiNii provides search functions for scholarly articles. Search result shows a list of articles corresponding to the query. CiNii also publishes "Bibliography Permalink", which is a web page describing the bibliography of every article stored in the database. It shows not only title and authors, but also the abstract and a list of references and citations. To become widely accepted by advanced users and developers, we are continuing to enhance CiNii, e.g., introducing permalinks and being the target of major search engines. In April 2009, CiNii redesigned and relaunched, and Linked Data (Berners-Lee, 2006) is offered for public use of scholarly information. In this paper we describe details of our Linked Data. FIG. 1. Screenshots of CiNii. Summary of Linked Data in CiNii is shown in Figure 2. -

Towards a Visual Annotation Tool for End-User Semantic Content Authoring
Towards a Visual Annotation Tool for End-User Semantic Content Authoring Torgeir Lebesbye and Ahmet Soylu Department of Informatics, University of Oslo, Norway {torgeirl, ahmets}@ifi.uio.no Abstract. There is a great amount of data on the Web, but to a large extent it is not published as linked data that computers can consume. Visual annotation tools have a considerable potential to empower end users to contribute their data to the Semantic Web, and could prove to be a solution to get more data on the Web linked. To this end, numerous tools have been developed; however, there still remains challenges to be addressed. In this paper, we present and discuss a set of prominent requirements toward the realisation of a visual annotation tool for end-user semantic content authoring. Keywords: Semantic Content Authoring, Direct Manipulation Interface, End-User Development 1 Introduction When Berners-Lee invented the Web in 1989, his motivation was to allow people to share and link documents without the barriers of hardware, file systems or data formats [4]. It later evolved into the Social Web, referred to as Web 2.0, where anyone could be the producers of contents through blogs, wikis and social media, and it became easy for people to collaborate on the Web. In later years, the introduction of semantic technologies has made it possible to describe the meaning of data in a language more consumable for computers: words written with markup languages like XML1, grammar structure using RDF2, and logic described in knowledge languages like OWL3 [7]. This means a contextual Web where data in documents are linked and can be mashed with data from other sources in a completely new way. -

Microformats.Cheatsheet
Elemental Microformats Design Patterns XFN VoteLinks Datetime Pattern <abbr rel="contact" rel="parent" rev="vote-for" microformats.org rel="acquaintance" rel="spouse" class="foo" hAtom hCalendar hCard hResume hReview rev="vote-against" • adr title="YYYY-MM-DDTHH:MM:SS+ZZ:ZZ" rel="friend" rel="kin" rev="vote-abstain" + country-name rel="met" rel="muse" >Human Date Time</abbr> • extended-address rel="co-worker" rel="crush" Rel-Nofollow + post-office-box rel="colleague" rel="date" rel="nofollow" Include Pattern + postal-code • street-address rel="co-resident" rel="sweetheart" Rel-Tag <object class="include" + locality rel="neighbor" rel="me" rel="tag" + region rel="child" type="text/html" • type Rel-Directory (Draft) data="#idref" • affiliation rel="directory" /> ¤ author + best bookmark (rel) Rel-License <a class="include" + + rel="license" + bday href="#idref">...</a> • • category + + class × contact Compound Microformats + description + dtend hCard hCalendar hResume (Draft) + dtreviewed × dtstart class="vcard" class="vevent" class="hresume" × dtstamp class="adr" class="category" rel="tag" class="affiliation" (hcard) duration class="type" class="class" class="education" (hcalendar) • education email [work|home|pref|postal|dom|intl] class="description" class="experience" (hcalendar) • • type class="post-office-box" class="dtend" (ISO Date) class="publication" (citation) • value class="street-address" class="dtstamp" (ISO Date) class="skill" rel="tag" • entry-content class="extended-address" class="dtstart" (ISO Date) class="summary" • entry-summary -

Using Microformats and Rdfa As Embeddable Data Formats
CHAPTER 18 Using Microformats and RDFa As Embeddable Data Formats The central problem that we will study in this chapter is how to embed information in web pages in a way that is easy to understand by both humans and computer programs. The solution that we will consider in depth is microformats, little chunks of structured data that are seamlessly embedded in web pages. (X)HTML is designed primarily to produce a human user interface (via a web browser). However, by carefully following certain conventions (ones that constitute microformats), you can produce (X)HTML out of which data can be unambiguously extracted. The consequence is that it is relatively easy to write computer programs to parse microformats so that the data can be reused in other contexts—giving rise to plenty of mashup possibilities. Moreover, having data embedded right in the context of a user interface is helpful. A user can decide what to do with this data (how to “operate” on a given piece of data) while in the context of normal browsing. Now, the previous paragraph is a bit abstract. What I will do in this chapter is walk you through some concrete examples describing microformats in general. We will use Operator, a Firefox extension, to help parse and view microformats and to create scripts that enable users to take specific actions in response to microformats. Specifically, we will do the following: * Study specific examples of microformats * Look at how to use the Firefox add-on Operator to jump-start your study of microformats * Look at programmatic approaches to consuming and creating microformats * Compare microformats to leading alternatives such as RDFa as a way to embed data in human-readable contexts.