Opsumit, INN-Macitentan
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Endothelin System and Therapeutic Application of Endothelin Receptor
xperim ACCESS Freely available online & E en OPEN l ta a l ic P in h l a C r m f o a c l a o n l o r g u y o J Journal of ISSN: 2161-1459 Clinical & Experimental Pharmacology Research Article Endothelin System and Therapeutic Application of Endothelin Receptor Antagonists Abebe Basazn Mekuria, Zemene Demelash Kifle*, Mohammedbrhan Abdelwuhab Department of Pharmacology, School of Pharmacy, College of Medicine and Health Sciences, University of Gondar, Gondar, Ethiopia ABSTRACT Endothelin is a 21 amino acid molecule endogenous potent vasoconstrictor peptide. Endothelin is synthesized in vascular endothelial and smooth muscle cells, as well as in neural, renal, pulmonic, and inflammatory cells. It acts through a seven transmembrane endothelin receptor A (ETA) and endothelin receptor B (ETB) receptors belongs to G protein-coupled rhodopsin-type receptor superfamily. This peptide involved in pathogenesis of cardiovascular disorder like (heart failure, arterial hypertension, myocardial infraction and atherosclerosis), renal failure, pulmonary arterial hypertension and it also involved in pathogenesis of cancer. Potentially endothelin receptor antagonist helps the treatment of the above disorder. Currently, there are a lot of trails both per-clinical and clinical on endothelin antagonist for various cardiovascular, pulmonary and cancer disorder. Some are approved by FAD for the treatment. These agents are including both selective and non-selective endothelin receptor antagonist (ETA/B). Currently, Bosentan, Ambrisentan, and Macitentan approved -

Comparison of Pharmacological Activity of Macitentan and Bosentan in Preclinical Models of Systemic and Pulmonary Hypertension
LFS-13929; No of Pages 7 Life Sciences xxx (2014) xxx–xxx Contents lists available at ScienceDirect Life Sciences journal homepage: www.elsevier.com/locate/lifescie Comparison of pharmacological activity of macitentan and bosentan in preclinical models of systemic and pulmonary hypertension Marc Iglarz ⁎, Alexandre Bossu, Daniel Wanner, Céline Bortolamiol, Markus Rey, Patrick Hess, Martine Clozel Drug Discovery Department, Actelion Pharmaceuticals Ltd, Gewerbestrasse 16, 4123 Allschwil, Switzerland article info abstract Article history: Aims: The endothelin (ET) system is a tissular system, as the production of ET isoforms is mostly autocrine or Received 29 October 2013 paracrine. Macitentan is a novel dual ETA/ETB receptor antagonist with enhanced tissue distribution and Accepted 12 February 2014 sustained receptor binding properties designed to achieve a more efficacious ET receptor blockade. To determine Available online xxxx if these features translate into improved efficacy in vivo, a study was designed in which rats with either systemic or pulmonary hypertension and equipped with telemetry were given macitentan on top of maximally effective Keywords: doses of another dual ET /ET receptor antagonist, bosentan, which does not display sustained receptor occupan- Endothelin A B Pharmacology cy and shows less tissue distribution. – Blood pressure Main methods: After establishing dose response curves of both compounds in conscious, hypertensive Dahl salt- Pulmonary hypertension sensitive and pulmonary hypertensive bleomycin-treated rats, macitentan was administered on top of the max- Rat imal effective dose of bosentan. Key findings: In hypertensive rats, macitentan 30 mg/kg further decreased mean arterial blood pressure (MAP) by 19 mm Hg when given on top of bosentan 100 mg/kg (n =9,p b 0.01 vs. -

Opsumit) Reference Number: CP.PHAR.194 Effective Date: 03.16 Last Review Date: 02.21 Line of Business: Medicaid Revision Log
Clinical Policy: Macitentan (Opsumit) Reference Number: CP.PHAR.194 Effective Date: 03.16 Last Review Date: 02.21 Line of Business: Medicaid Revision Log See Important Reminder at the end of this policy for important regulatory and legal information. Description Macitentan (Opsumit®) is an endothelin receptor antagonist. FDA Approved Indication(s) Opsumit is indicated for treatment of pulmonary arterial hypertension (PAH) (World Health Organization (WHO) Group I) to reduce the risks of disease progression and hospitalization for PAH. Effectiveness was established in a long-term study in PAH patients with predominantly WHO Functional Class II-III symptoms treated for an average of 2 years. Patients had idiopathic and heritable PAH (57%), PAH caused by connective tissue disorders (31%), and PAH caused by congenital heart disease with repaired shunts (8%). Policy/Criteria Provider must submit documentation (such as office chart notes, lab results or other clinical information) supporting that member has met all approval criteria. It is the policy of health plans affiliated with Centene Corporation® that Opsumit is medically necessary when the following criteria are met: I. Initial Approval Criteria A. Pulmonary Arterial Hypertension (must meet all): 1. Diagnosis of PAH; 2. Prescribed by or in consultation with a cardiologist or pulmonologist; 3. Failure of a calcium channel blocker (see Appendix B), unless member meets one of the following (a or b): a. Inadequate response or contraindication to acute vasodilator testing; b. Contraindication or clinically significant adverse effects to calcium channel blockers are experienced; 4. Failure of generic ambrisentan or bosentan, unless clinically significant adverse effects are experienced or both are contraindicated; 5. -

Doctors Healthcare Plans, Inc. Is an HMO with a Medicare Contract
ACTEMRA IV (S) MEDICATION(S) ACTEMRA 200 MG/10 ML VIAL, ACTEMRA 400 MG/20 ML VIAL, ACTEMRA 80 MG/4 ML VIAL COVERED USES All medically accepted indications not otherwise excluded from Part D. EXCLUSION CRITERIA N/A REQUIRED MEDICAL INFORMATION Rheumatoid Arthritis (RA) (Initial): Diagnosis of moderately to severely active RA. One of the following: Failure, contraindication, or intolerance to both Enbrel (etanercept) and Humira (adalimumab), OR for continuation of prior Actemra therapy. Systemic Juvenile Idiopathic arthritis (SJIA) (Initial): Diagnosis of active SJIA. Failure, contraindication, or intolerance to one NSAID or systemic glucocorticoid. Polyarticular Juvenile Idiopathic Arthritis (PJIA) (Initial): Diagnosis of active PJIA. One of the following: Failure, contraindication, or intolerance to both Enbrel (etanercept) and Humira (adalimumab), OR for continuation of prior Actemra therapy. All indications (Initial, reauth): Patient is not receiving Actemra in combination with a biologic DMARD [eg, Enbrel (etanercept), Humira (adalimumab), Cimzia (certolizumab), Simponi (golimumab)]. AGE RESTRICTION N/A PRESCRIBER RESTRICTION All uses (Initial): Prescribed by or in consultation with a rheumatologist. COVERAGE DURATION All uses (Initial, reauth): 12 months OTHER CRITERIA All uses (Reauth): Documentation of positive clinical response to Actemra therapy. PAGE 1 LAST UPDATED 12/2019 ACTEMRA SC MEDICATION(S) ACTEMRA 162 MG/0.9 ML SYRINGE, ACTEMRA ACTPEN COVERED USES All medically accepted indications not otherwise excluded from Part D. EXCLUSION CRITERIA N/A REQUIRED MEDICAL INFORMATION Rheumatoid Arthritis (RA) (Initial): Diagnosis of moderately to severely active RA. One of the following: Trial and failure, contraindication, or intolerance to both Enbrel (etanercept) and Humira (adalimumab), OR for continuation of prior Actemra therapy. -

2020 Aetna Standard Plan
Plan for your best health Aetna Standard Plan Aetna.com Aetna is the brand name used for products and services provided by one or more of the Aetna group of subsidiary companies, including Aetna Life Insurance Company and its affiliates (Aetna). Aetna Pharmacy Management refers to an internal business unit of Aetna Health Management, LLC. Aetna Pharmacy Management administers, but does not offer, insure or otherwise underwrite the prescription drug benefits portion of your health plan and has no financial responsibility therefor. 2020 Pharmacy Drug Guide - Aetna Standard Plan Table of Contents INFORMATIONAL SECTION..................................................................................................................6 *ADHD/ANTI-NARCOLEPSY/ANTI-OBESITY/ANOREXIANTS* - DRUGS FOR THE NERVOUS SYSTEM.................................................................................................................................16 *ALLERGENIC EXTRACTS/BIOLOGICALS MISC* - BIOLOGICAL AGENTS...............................18 *ALTERNATIVE MEDICINES* - VITAMINS AND MINERALS....................................................... 19 *AMEBICIDES* - DRUGS FOR INFECTIONS.....................................................................................19 *AMINOGLYCOSIDES* - DRUGS FOR INFECTIONS.......................................................................19 *ANALGESICS - ANTI-INFLAMMATORY* - DRUGS FOR PAIN AND FEVER............................19 *ANALGESICS - NONNARCOTIC* - DRUGS FOR PAIN AND FEVER......................................... -
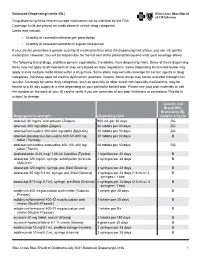
Balanced Drug List Dispensing Limits
Balanced Dispensing Limits (DL) Drug dispensing limits help encourage medication use as intended by the FDA. Coverage limits are placed on medications in certain drug categories. Limits may include: • Quantity of covered medication per prescription • Quantity of covered medication in a given time period If your doctor prescribes a greater quantity of medication than what the dispensing limit allows, you can still get the medication. However, you will be responsible for the full cost of the prescription beyond what your coverage allows. The following brand drugs, and their generic equivalents, if available, have dispensing limits. Some of these dispensing limits may not apply to all members or may vary based on state regulations. Some dispensing limits listed below may apply across multiple medications within a drug class. Some plans may exclude coverage for certain agents or drug categories, like those used for erectile dysfunction (example: Viagra). Some drugs may not be available through mail service. Coverage for some drug categories, such as specialty or other select non‑specialty medications, may be limited to a 30‑day supply at a time depending on your particular benefit plan. Please see your plan materials or call the number on the back of your ID card to verify if you are uncertain of any plan limitations or exclusions. This list is subject to change. Generic and Brand (BG), Brand Only (B), Drug (generic) strength Dispensing Limit Generic only (G) abacavir 20 mg/mL oral solution (Ziagen) 960 mL per 30 days BG abacavir 300 mg tablet -

OPSUMIT (Macitentan) Is an Endothelin Receptor Antagonist
HIGHLIGHTS OF PRESCRIBING INFORMATION -------------------------CONTRAINDICATIONS------------------------ These highlights do not include all the information needed to use • Pregnancy (4.1) OPSUMIT safely and effectively. See full prescribing information for OPSUMIT. -------------------WARNINGS AND PRECAUTIONS--------------- • ERAs cause hepatotoxicity and liver failure. Obtain baseline liver OPSUMIT (macitentan) tablets, for oral use enzymes and monitor as clinically indicated (5.3). Initial U.S. Approval: 2013 • Fluid retention may require intervention (5.4) • Decreases in hemoglobin (5.5). WARNING: EMBRYO-FETAL TOXICITY • See full prescribing information for complete boxed warning Pulmonary edema in patients with pulmonary veno-occlusive disease. If confirmed, discontinue treatment (5.6). • Do not administer OPSUMIT to a pregnant female because • it may cause fetal harm (4.1, 5.1, 8.1). Decreases in sperm count have been observed in patients taking • Females of reproductive potential: exclude pregnancy before ERAs (5.7). start of treatment, monthly during treatment, and 1 month -------------------------ADVERSE REACTIONS------------------------- after stopping treatment. Prevent pregnancy during Most common adverse reactions (more frequent than placebo by ≥3%) treatment and for one month after treatment by using are anemia, nasopharyngitis/pharyngitis, bronchitis, headache, acceptable methods of contraception (2.2, 8.3). influenza, and urinary tract infection (6.1). • For all female patients, OPSUMIT is available only through a restricted -

Ambrisentan (Letairis and Opsumit)
US Family Health Plan Prior Authorization Request Form for ambrisentan (Letairis), macitentan (Opsumit) To be completed and signed by the prescriber. To be used only for prescriptions which are to be filled through the Department of Defense (DoD) US Family Health Plan Pharmacy Program. US Family Health Plan is a TRICARE contractor for DoD. The completed form may be faxed to 855-273-5735 OR The patient may attach the completed form to the prescription and mail it to: Attn: Pharmacy, 77 Warren St, Brighton, MA 02135 QUESTIONS? Call 1-877-880-7007 1B Step 3BPlease complete patient and physician information (please print): 2B1 Patient Name: Physician Name: Address: Address: Sponsor ID # Phone #: Date of Birth: Secure Fax #: 4B Step 6BPlease complete the clinical assessment: 5B2 1. 7BDoes the patient have a documented diagnosis of WHO Yes No group 1 PAH? Proceed to question 2 STOP Cov erage not approv ed 2. 8BIs the requested medication being prescribed by or in Yes No consultation with a cardiologist or a pulmonologist? Proceed to question 3 STOP Cov erage not approv ed 3. 9BHas the patient had a right heart catheterization? Yes No Proceed to question 4 STOP Cov erage not approv ed 4. 10BIs documentation being provided to confirm that the patient Yes No has had a right heart catheterization? Proceed to question 5 STOP PLEASE NOTE: Medical documentation specific to your Cov erage not approv ed response to this question must be attached to this case or your request could be denied. Documentation may include, but is not limited to, chart notes and catheterization laboratory reports. -

Bosentan Or Macitentan Therapy in Chronic Thromboembolic Pulmonary Hypertension?
Lung (2019) 197:753–760 https://doi.org/10.1007/s00408-019-00274-9 PULMONARY HYPERTENSION Bosentan or Macitentan Therapy in Chronic Thromboembolic Pulmonary Hypertension? M. C. J. van Thor1,2 · L. ten Klooster2 · R. J. Snijder2 · J. C. Kelder3 · J. J. Mager2 · M. C. Post1 Received: 28 June 2019 / Accepted: 19 September 2019 / Published online: 3 October 2019 © Springer Science+Business Media, LLC, part of Springer Nature 2019 Abstract Objective Research comparing bosentan and macitentan in chronic thromboembolic pulmonary hypertension (CTEPH) is scarce, although macitentan might have superior pharmacologic properties. We present the frst real-world, 2-year follow-up results and compare clinical outcomes of both drugs in CTEPH. Methods All consecutive, technical inoperable or residual CTEPH patients receiving bosentan or macitentan, diagnosed in our multidisciplinary team between January 2003 and January 2019, were included. We report and compare survival, clini- cal worsening (CW), adverse events, WHO FC, NT-proBNP and 6-min walking test (6MWT) until 2 years after medication initiation. Results In total, 112 patients receiving bosentan or macitentan (58% female, mean age 62 ± 14 years, 68% WHO FC III/IV, 51% bosentan) could be included. Mean treatment duration was 1.9 ± 0.4 years for bosentan and 1.2 ± 0.6 years for macitentan. Two-year survival rate was 91% for bosentan and 80% for macitentan (HR mortality macitentan 1.85 [0.56–6.10], p = 0.31). Two-year CW-free survival was 81% and 58%, respectively (HR CW macitentan 2.16 [0.962–4.87], p = 0.06). Right atrial pressure, cardiac output (for mortality alone) and 6MWT lowest saturation were multivariate predictors at baseline. -

Perioperative Medication Management - Adult/Pediatric - Inpatient/Ambulatory Clinical Practice Guideline
Effective 6/11/2020. Contact [email protected] for previous versions. Perioperative Medication Management - Adult/Pediatric - Inpatient/Ambulatory Clinical Practice Guideline Note: Active Table of Contents – Click to follow link INTRODUCTION........................................................................................................................... 3 SCOPE....................................................................................................................................... 3 DEFINITIONS .............................................................................................................................. 3 RECOMMENDATIONS ................................................................................................................... 4 METHODOLOGY .........................................................................................................................28 COLLATERAL TOOLS & RESOURCES..................................................................................................31 APPENDIX A: PERIOPERATIVE MEDICATION MANAGEMENT .................................................................32 APPENDIX B: TREATMENT ALGORITHM FOR THE TIMING OF ELECTIVE NONCARDIAC SURGERY IN PATIENTS WITH CORONARY STENTS .....................................................................................................................58 APPENDIX C: METHYLENE BLUE AND SEROTONIN SYNDROME ...............................................................59 APPENDIX D: AMINOLEVULINIC ACID AND PHOTOTOXICITY -

Bluecare, Bluedirect, Blueessential, Bluepartner, Blueprime and Simplyblue Formularies Quantity Limit Program Summary
BCBSND BlueCare, BlueDirect, BlueEssential, BluePartner, BluePrime and SimplyBlue Formularies Quantity Limit Program Summary Quantity Limit Program Summary Opioid Immediate Release (Opioids IR) target drugs may be subject to quantity limits according to the Opioid Immediate Release (IR) New to Therapy Program https://www.myprime.com/content/dam/prime/memberportal/forms/AuthorForms/BCBSND/Commercial/ND_Opioids_IR_NTT_QL_ProgSum.pdf Quantity Limits To help with safe and effective drug use, some drugs have limits on how much of a particular drug you can get for a specific time period. If there is medical reason why you need an amount that is greater than what is allowed, your physician can fill out a Quantity Limit prior authorization form for you and send it to Prime Therapeutics. This form can be found on the MyPrime.com website. Note: Quantity limits apply to all available MultiSource Code (MSC) products Your health benefit plan may not cover certain prescription drug products or drug categories included in this document. Please consult your benefit plan materials for details about your particular benefit. This document may include drugs that are not included on your plan's formulary. For drug coverage status, please consult your plan's formulary. QL PROG NAME Target GPI Name Quantity Limit Acute Migraine Agents NURTEC RIMEGEPANT SULFATE TAB DISINT 75 MG 45 Tablets Per 90 DAYS Acute Migraine Agents REYVOW LASMIDITAN SUCCINATE TAB 100 MG 8 Tablets Per 30 DAYS Acute Migraine Agents REYVOW LASMIDITAN SUCCINATE TAB 50 MG 8 Tablets Per 30 -

Minutes of the CHMP Meeting 25-29 January 2021
04 March 2021 EMA/CHMP/128746/2021 Human Medicines Division Committee for medicinal products for human use (CHMP) Minutes for the meeting on 25-29 January 2021 Chair: Harald Enzmann – Vice-Chair: Bruno Sepodes Disclaimers Some of the information contained in this set of minutes is considered commercially confidential or sensitive and therefore not disclosed. With regard to intended therapeutic indications or procedure scopes listed against products, it must be noted that these may not reflect the full wording proposed by applicants and may also vary during the course of the review. Additional details on some of these procedures will be published in the CHMP meeting highlights once the procedures are finalised and start of referrals will also be available. Of note, these minutes are a working document primarily designed for CHMP members and the work the Committee undertakes. Note on access to documents Some documents mentioned in the minutes cannot be released at present following a request for access to documents within the framework of Regulation (EC) No 1049/2001 as they are subject to on-going procedures for which a final decision has not yet been adopted. They will become public when adopted or considered public according to the principles stated in the Agency policy on access to documents (EMA/127362/2006). Official address Domenico Scarlattilaan 6 ● 1083 HS Amsterdam ● The Netherlands Address for visits and deliveries Refer to www.ema.europa.eu/how-to-find-us Send us a question Go to www.ema.europa.eu/contact Telephone +31 (0)88 781 6000 An agency of the European Union © European Medicines Agency, 2021.