Mechanisms of Gabaergic Homeostatic Plasticity Peter Wenner, Emory University
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

IT Reprint Hindawi
Reprint and OA content. Emboldened by the suc- role in establishing the Open Access Hindawi cess of the hybrid model, Hindawi moved Scholarly Publishers Association (OASPA; to an all-OA approach for its journals by www.oaspa.org). According to OASPA, the 2007. By the end of that year, the company group’s aim is to support and “represent Publishing: announced a joint initiative with SAGE the interests of Open Access (OA) journal Publications to publish a collection of OA publishers globally in all scientific, tech- journals, leveraging Hindawi’s production nical, and scholarly disciplines … through A Working technology and SAGE’s editorial expertise. exchanging information, setting stan- dards, advancing models, advocacy, edu- OA Model Keys to Rapid Growth cation, and the promotion of innovation.” The move to an all-OA model has been Who’s the Customer? paying off. Paul Peters, Hindawi’s head by NANCY DAVIS KHO | of business development, says the com- In traditional scholarly publishing, the pany now publishes 275 journals, about customer has been a library subscriber to In the 3 years since academic pub- 10% of which are carried on the SAGE- journals. “We were terrible at dealing with lisher Hindawi Publishing Corp. moved Hindawi Access to Research platform. libraries,” says Peters. “Being in Egypt, to a strictly open access (OA) publishing Reflecting the growth in the number you just can’t send a sales team over to model, the perception of OA has shifted. of journals published is a commensurate talk to them about our journal offerings.” Once viewed as a radical upending of tra- jump in submissions. -

Gold Route Open Access Journals in Engineering and Technology: Analysis of Research Impact and Implications for Scholarly Communication
Gold Route Open Access Journals in Engineering and Technology: Analysis of Research Impact and Implications for Scholarly Communication Ifeanyi J. Ezema, Ph.D Research Fellow Department of Information Science University of South Africa Nnamdi Azikiwe Library University of Nigeria Nsukka [email protected] Abstract This study examines the research impact of gold open access (OA) journals in engineering and technology. A total of 37 gold route journals that request article processing charges (APC) and 66 other OA journals without APC (i.e., platinum OA) in the field were found in the Directory of Open Access Journals and used for the study. The publishers, APC charges, and the year each journal was added to DOAJ were identified and included in this study. An informetric approach was used to extract research impact indicators (citations, paper productivity, cite/paper, cite/journal, and h-index) of journals. Findings revealed that the publishers of the APC journals were dominated by commercial publishing companies, producing over 60% of the journals. Article processing charges ranged from $7.60 to $3,471.50, with an average of $727.00. Gold route open access (APC) journals performed better than open access non-APC OA journals, since they produced 51.2% of the papers, 71.8% of the total citations, and 65.1% of the total h- index. There was a weak positive correlation between the APC amount of the gold route journals and their research impact. Introduction The past one and a half decades has witnessed a revolution in scholarly communication with the emergence of open access publishing. -

Working Together to Maximize Efficiency During the COVID-19 Pandemic
Working together to maximize efficiency during the COVID-19 pandemic A cross-publisher initiative #C19RapidReview Last updated: August 2020 This cross-publisher collaboration endorsed by the Open Access Scholarly Publishers Association (OASPA) aims to ensure research related to COVID-19 is reviewed and published as quickly as possible. We have come together across the open publishing industry to support academics; peer reviewers, editors and authors, and to ensure the widest and quickest dissemination of relevant quality content. To this end, we encourage academics to sign up to a reviewer database. Please, consider signing up as a rapid reviewer by filling in this form. We also encourage authors to use preprint servers and call on other publishers to action with a focus on open data. Read the formal Letter of Intent for more information and help us spread the word on social media using #C19RapidReview. List of organizations and journal titles participating in the #C19RapidReview initiative Royal Society Proceedings A Proceedings B Open Biology Biology Letters Interface Royal Society Open Science Hindawi Advances in Public Health Analytical Cellular Pathology BioMed Research International Canadian Journal of Infectious Diseases and Medical Microbiology Canadian Respiratory Journal Cardiology Research and Practice Case Reports in Critical Care Case Reports in Emergency Medicine Case Reports in Immunology Case Reports in Infectious Diseases Case Reports in Medicine Case Reports in Otolaryngology Case Reports in Pulmonology Complexity -

Abbreviations of Names of Serials
Abbreviations of Names of Serials This list gives the form of references used in Mathematical Reviews (MR). ∗ not previously listed The abbreviation is followed by the complete title, the place of publication x journal indexed cover-to-cover and other pertinent information. y monographic series Update date: January 30, 2018 4OR 4OR. A Quarterly Journal of Operations Research. Springer, Berlin. ISSN xActa Math. Appl. Sin. Engl. Ser. Acta Mathematicae Applicatae Sinica. English 1619-4500. Series. Springer, Heidelberg. ISSN 0168-9673. y 30o Col´oq.Bras. Mat. 30o Col´oquioBrasileiro de Matem´atica. [30th Brazilian xActa Math. Hungar. Acta Mathematica Hungarica. Akad. Kiad´o,Budapest. Mathematics Colloquium] Inst. Nac. Mat. Pura Apl. (IMPA), Rio de Janeiro. ISSN 0236-5294. y Aastaraam. Eesti Mat. Selts Aastaraamat. Eesti Matemaatika Selts. [Annual. xActa Math. Sci. Ser. A Chin. Ed. Acta Mathematica Scientia. Series A. Shuxue Estonian Mathematical Society] Eesti Mat. Selts, Tartu. ISSN 1406-4316. Wuli Xuebao. Chinese Edition. Kexue Chubanshe (Science Press), Beijing. ISSN y Abel Symp. Abel Symposia. Springer, Heidelberg. ISSN 2193-2808. 1003-3998. y Abh. Akad. Wiss. G¨ottingenNeue Folge Abhandlungen der Akademie der xActa Math. Sci. Ser. B Engl. Ed. Acta Mathematica Scientia. Series B. English Wissenschaften zu G¨ottingen.Neue Folge. [Papers of the Academy of Sciences Edition. Sci. Press Beijing, Beijing. ISSN 0252-9602. in G¨ottingen.New Series] De Gruyter/Akademie Forschung, Berlin. ISSN 0930- xActa Math. Sin. (Engl. Ser.) Acta Mathematica Sinica (English Series). 4304. Springer, Berlin. ISSN 1439-8516. y Abh. Akad. Wiss. Hamburg Abhandlungen der Akademie der Wissenschaften xActa Math. Sinica (Chin. Ser.) Acta Mathematica Sinica. -

Working Together to Maximize Efficiency During the COVID-19 Pandemic
Working together to maximize efficiency during the COVID-19 pandemic A cross-publisher initiative #C19RapidReview Last updated: May 2020 This cross-publisher collaboration endorsed by the Open Access Scholarly Publishers Association (OASPA) aims to ensure research related to COVID-19 is reviewed and published as quickly as possible. We have come together across the open publishing industry to support academics; peer reviewers, editors and authors, and to ensure the widest and quickest dissemination of relevant quality content. To this end, we encourage academics to sign up to a reviewer database. Please, consider signing up as a rapid reviewer by filling in this form. We also encourage authors to use preprint servers and call on other publishers to action with a focus on open data. Read the formal Letter of Intent for more information and help us spread the word on social media using #C19RapidReview. List of organizations and journal titles participating in the #C19RapidReview initiative Royal Society Proceedings A Proceedings B Open Biology Biology Letters Interface Royal Society Open Science Hindawi Advances in Public Health Analytical Cellular Pathology BioMed Research International Canadian Journal of Infectious Diseases and Medical Microbiology Canadian Respiratory Journal Cardiology Research and Practice Case Reports in Critical Care Case Reports in Emergency Medicine Case Reports in Immunology Case Reports in Infectious Diseases Case Reports in Medicine Case Reports in Otolaryngology Case Reports in Pulmonology Complexity -

Definitions of Predatory Publishing (E.G., Clark and Smith, 2015; Grudniewicz Et Al., 2019)
1 The concept of predatory publishing is complex and often controversial. There are numerous – often similar – definitions of predatory publishing (e.g., Clark and Smith, 2015; Grudniewicz et al., 2019). However, these definitions are broad and rely on subjective, normative judgments of best practices and professional ethics in academic publishing. In part due to the subjectivity involved in many definitions of predatory publishing, debates about predatory publishing are often highly contentious. Different scholars and academic stakeholders may have different philosophies and beliefs regarding the appropriateness and legitimacy of varying publishing practices. Our research provides empirical data on non-indexed publishers to enable scholars and academic stakeholders to make informed decisions about the legitimacy – or lack thereof – of journals and publishers, regardless of professional preferences and philosophies. The opacity of peer review in most academic journals makes it difficult to directly observe the legitimacy and quality of peer review. Although manuscript development and gatekeeping processes vary between different journals, it is often difficult to know what happens in the “black box” of peer review. This challenge is compounded with predatory or questionable academic journals, as such publishers often operate in covert and/or deceptive manners. Further, predatory journals and publishers are rarely indexed by institutions like the Web of Science, which makes surveilling and analyzing such journals difficult. However, document data and metadata can provide empirical evidence about the professionalism and operating procedures of questionable academic journals. Using a variety of web scraping techniques, we developed a database of non-indexed academic publishers, which includes numerous publishers on the Cabells Predatory Reports list. -

Hindawi Publishing Corporation: the Open Access Contributor Pays Model Cairo, Egypt
Hindawi Publishing Corporation: The Open Access Contributor Pays Model Cairo, Egypt www.hindawi.com JISC Content Ithaka Case Studies in Sustainability Hindawi Publishing Corporation, a Cairo-based for- profit publisher of journals in science, technology and medicine, was founded as a subscription-based publisher in 1997. By 2003, Hindawi had begun exploring Open Access (OA) models, and by 2007 had become an entirely OA publisher; its current list consists of 160 OA journals. Hindawi’s financial model is based on charging contributors a fee per article published, a model also currently used by BioMed Central and the Public Library of Science (PLoS), among others. Since 2007 Hindawi has continued to refine its business model, in particular through its partnership with the scholarly publisher SAGE and by introducing institutional memberships earlier this year. This case study will explore Hindawi’s path to choosing this financial model, the opportunities this choice has offered and the challenges it has posed. Introduction Hindawi Publishing Corporation began in 1997 as a small scholarly publisher of subscription-based peer-reviewed online journals. Its founders, Ahmed Hindawi and Nagwa Abdel- Mottaleb, were motivated to start their company because of their belief that the ‘low cost base and access to a large workforce of ambitious, well-educated young professionals’ in Cairo would allow them to offer ‘a labour-intensive service at a level that is not economically viable for most publishers based in the West’.1 In particular, they built their business by stressing the administrative and editorial support they could offer their editors and referees, the strength of their custom-built web-based submission and publication platform, and the efficiency of their highly automated production process. -

Hindawi Slide Template
Building a radically open future Catriona J. MacCallum Director of Open Science, Hindawi Barcelona June 2018 ORCID 0000-0001-9623-2225 @Hindawi @catmacOA Open Access since 2007 Free access – no charge to access ~18,000 peer-reviewed articles a year No embargos – immediately available Science, Technology & Medicine Reuse – Creative Commons Attribution License (CC BY) - use with proper attribution A founding member of OASPA September 2017 November 2016 Proportion of Open Access (Crossref) •Gold: Published in an open-access journal (as defined by the DOAJ). •Green: Toll-access on the publisher page, but there is a free copy in an OA repository. •Hybrid: Free under an open license in a toll-access journal. •Bronze: Free to read on the publisher page, but without a license. •Closed: All other articles, including those shared only on an ASN or in Sci-Hub CC BY Piwowar H, Priem J, Larivière V, Alperin JP, Matthias L, Norlander B, Farley A, West J, Haustein S. (2017) The State of OA: A large-scale analysis of the prevalence and impact of Open Access articles. PeerJ Preprints 5:e3119v1 https://doi.org/10.7287/peerj.preprints.3119v1 Open Access by publisher CC BY Piwowar H, Priem J, Larivière V, Alperin JP, Matthias L, Norlander B, Farley A, West J, Haustein S. (2017) The State of OA: A large-scale analysis of the prevalence and impact of Open Access articles. PeerJ Preprints 5:e3119v1 https://doi.org/10.7287/peerj.preprints.3119v1 Open Access by discipline CC BY Piwowar H, Priem J, Larivière V, Alperin JP, Matthias L, Norlander B, Farley A, West J, Haustein S. -

Alleged Predatory Publisher Buys Medical Journals
News CMAJ Alleged predatory publisher buys medical journals ne of the world’s most well- known “predatory” publishers O has bought two commercial Canadian publishers of about 16 medi- cal specialty journals. But one former owner says during the purchasing negotiations, the new publisher agreed not use predatory practices. The journals were previously pub- lished by Pulsus Group in Oakville, Ontario, and Andrew John Publishing in Dundas, Ontario. As of September 28, the journals appear on the Pulsus Group website, which gives an address in Lon- don, UK, where Pulsus Group UK was registered in August 2016. It is a subsid- Pulsus Group iary of OMICS International of Hyder- Pulsus journals are now owned by a newly created subsidiary of OMICS International, abad, India, which is facing charges from an alleged predatory publisher based in Hyderabad, India. the US Federal Trade Commission (FTC) for “deceiving academics and researchers OMICS. Brockington learned this during nals’ impact factors and listing in Scopus about the nature of its publications and a conference call with the new publisher and PubMed, in order to look legitimate, hiding publication fees ranging from hun- when she asked who now owned Pulsus he adds. dreds to thousands of dollars,” according Group. When she contacted editors of Beall says OMICS’ main practice is to an FTC statement. other Pulsus journals, they were unaware to “hold papers hostage.” Authors sub- Prior to this latest sale, back in that the new publisher was OMICS. mit papers in response to spam emails December 2015, Pulsus sold four jour- When Robert Kalina decided to looking for submissions. -
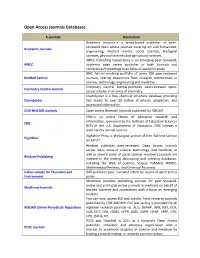
Open Access Journals Databases
Open Access Journals Databases E-journals Description Academic Journals is a broad-based publisher of peer- reviewed open access journals covering art and humanities, Academic Journals engineering, medical science, social sciences, biological sciences, physical sciences and agricultural sciences. AIRCC Publishing Corporation is an emerging peer-reviewed, AIRCC academic open access publisher of both Journals and conference Proceedings in all fields of academic study. BMC has an evolving portfolio of some 300 peer-reviewed BioMed Central journals, sharing discoveries from research communities in science, technology, engineering and medicine. Chemistry Central Journal publishes peer-reviewed open- Chemistry Central Journal access articles in all areas of chemistry. ChemSpider is a free chemical structure database providing ChemSpider fast access to over 58 million structures, properties, and associated information. CSIR-NISCAIR Journals Open access Research Journals published by NISCAIR ERIC is an online library of education research and information, sponsored by the Institute of Education Sciences ERIC (IES) of the U.S. Department of Education. ERIC indexes a wide variety journal sources. HighWire Press is the largest archive of free full-text science HighWire on Earth! Hindawi publishes peer-reviewed, Open Access journals across many areas of science, technology, and medicine, as well as several areas of social science. Hindawi’s journals are Hindawi Publishing indexed in the leading abstracting and indexing databases, including the Web of Science, Scopus, PubMed, INSPEC, Mathematical Reviews, and Chemical Abstracts. Indian society for Education and ISEE publishes peer reviewed article by means of open access Environment policy. Medknow provides publishing services for peer-reviewed, online and print-plus-online journals in medicine on behalf of Medknow Journals learned societies and associations with a focus on emerging markets. -

Everything You Ever Wanted to Know About Predatory Publishing but Were Afraid to Ask
Everything You Ever Wanted to Know About Predatory Publishing but Were Afraid to Ask Monica Berger* Introduction: Librarians and Predatory Publishing Librarians have a key role to play in educating users about predatory publishing. Predatory publishing can be described as low quality, amateurish, and often unethical academic publishing that is usually Open Access (OA). Understanding predatory publishing helps authors to make more informed decisions about where to publish. In the process of educating our users, librarians can set the ground for important conversations that encourage critical thinking about the scholarly communications process. Predatory publishing stems from broader prob- lems including overemphasis on publication quantity, an OA models based on traditional, for-profit publishing, and resource disparities in the Global South. When users take fuller responsibility and ownership of scholarly communications, knowledge can be a public good and not a commodity. A more sustainable and just scholarly communications ecosystem can be a reality. As effective advocates for OA, librarians need to be ready to respond to those who conflate OA and preda- tory publishing. It is helpful to contextualize predatory publishing as an aspect of evaluating publishers and the quality of scholarship. This helps promote the idea that due diligence is the responsibility of all scholars, whether as authors, peers, or administrators. Additionally, positioning (deliberate) predatory publishing in the broader arena of unethical and fraudulent scholarly practices helps to decouple predatory publishing from OA and boosts our ability to communicate effectively with non-librarians. Overview Defining predatory publishing is challenging: the word “predatory” may not do justice to a complex subject. -

Open Access Journals & Article Processing Charges 2011
Open access journals & article processing charges 2011 – 2021. Preprint. June 23, 2021. 1 Open access journals & article processing charges 2011 - 2021 Principal Investigator & Corresponding author: Heather Morrison [email protected] School of Information Studies, University oF Ottawa 111-08 Desmarais, 55 Laurier Avenue East Ottawa, Ontario, Canada K1N 6N5 Authors Luan Borges, University of Ottawa [email protected] Xuan Zhao, University of Ottawa [email protected] Tanoh Laurent Kakou, University oF Ottawa [email protected] Amit Nataraj Shanbhoug, University of Ottawa [email protected] Biographical inFormation about the authors and additional members of the Sustaining the Knowledge Commons team: https://sustainingknowledgecommons.org/about-the-team/ Open access journals & article processing charges 2011 – 2021. Preprint. June 23, 2021. 2 Abstract This study examines trends in open access article processing charges (APCs) from 2011 – 2021, building on a 2011 study by Solomon & Björk (2012). Two methods are employed, a modified replica and a status update of the 2011 journals. Data is drawn From multiple sources and datasets are available as open data (Morrison et al, 2021). Most journals do not charge APCs; this has not changed. The global average per-journal APC increased slightly, from 906 USD to 958 USD, while the per-article average increased from 904 USD to 1,626 USD, indicating that authors choose to publish in more expensive journals. Publisher siZe, type, impact metrics and subject afFect charging tendencies, average APC and pricing trends. About halF the journals From the 2011 sample are no longer listed in DOAJ in 2021, due to ceased publication or publisher de-listing.