Ep 2611776 B1
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Carboxylic Acid Functionalized Butyl Rubber: from Synthesis to Applications
Western University Scholarship@Western Electronic Thesis and Dissertation Repository 2-7-2013 12:00 AM Carboxylic Acid Functionalized Butyl Rubber: From Synthesis to Applications Matthew John McEachran The University of Western Ontario Supervisor Dr. Elizabeth R. Gillies The University of Western Ontario Graduate Program in Chemistry A thesis submitted in partial fulfillment of the equirr ements for the degree in Master of Science © Matthew John McEachran 2013 Follow this and additional works at: https://ir.lib.uwo.ca/etd Part of the Materials Chemistry Commons, Medicinal-Pharmaceutical Chemistry Commons, and the Polymer Chemistry Commons Recommended Citation McEachran, Matthew John, "Carboxylic Acid Functionalized Butyl Rubber: From Synthesis to Applications" (2013). Electronic Thesis and Dissertation Repository. 1117. https://ir.lib.uwo.ca/etd/1117 This Dissertation/Thesis is brought to you for free and open access by Scholarship@Western. It has been accepted for inclusion in Electronic Thesis and Dissertation Repository by an authorized administrator of Scholarship@Western. For more information, please contact [email protected]. Carboxylic Acid Functionalized Butyl Rubber: From Synthesis to Applications (Spine title: Carboxylic Acid Functionalized Butyl Rubber) (Thesis format: Integrated Article) by Matthew John McEachran Graduate Program in Chemistry A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science The School of Graduate and Postdoctoral Studies The University of Western Ontario London, -

Chem 341 • Organic Chemistry I Lecture Summary 07 • September 07, 2007
Chem 341 • Organic Chemistry I Lecture Summary 07 • September 07, 2007 Chapter 2 - Polar Covalent Bonds; Acids and Bases Alkyl Substituents When an alkane is a smaller part of a larger molecule (a substituent), it is called an ‘alkyl’ group. The parent name for the number of carbons remains the same, but it is given a ‘yl’ ending indicating it is a sub-part of the molecule. See for example several common names for some organic molecules shown below. Note that for chlorobutane, there are four different constitutional isomers possible. Normal butane refers to the straight chain. If the chlorine is moved to a secondary carbin it is called a secondary chloride. If the carbon skeleton is changed, it becomes an iso-butyl group and if the chlorine is attached to the tertiary carbon it is a tert-butyl group. These common names are still in use, though there is a more exact systematic naming system that is easier to understand (but longer names). H3C Cl OH methyl chloride ethyl alcohol Cl Cl Cl Cl n-butyl chloride sec-butyl chloride iso-butyl chloride tert-butyl chloride 1-chlorobutane 2-chlorobutane 1-chloro-2-methylpropane 2-chloro-2-methylpropane Degree of Alkyl Substitution We designate a kind of carbon (or kind of functional group attached to that carbon) according to how many other alkyl groups are attached to it. For example, a carbon on the end of a chain (with one alkyl group) would be a primary carbon. The hydrogens attached to that carbon would be called primary hydrogens. An alcohol attached to a primary carbon would be called a primary alcohol. -

Chapter 16 the Chemistry of Benzene and Its Derivatives
Instructor Supplemental Solutions to Problems © 2010 Roberts and Company Publishers Chapter 16 The Chemistry of Benzene and Its Derivatives Solutions to In-Text Problems 16.1 (b) o-Diethylbenzene or 1,2-diethylbenzene (d) 2,4-Dichlorophenol (f) Benzylbenzene or (phenylmethyl)benzene (also commonly called diphenylmethane) 16.2 (b) (d) (f) (h) 16.3 Add about 25 °C per carbon relative to toluene (110.6 C; see text p. 743): (b) propylbenzene: 161 °C (actual: 159 °C) 16.4 The aromatic compound has NMR absorptions with greater chemical shift in each case because of the ring current (Fig. 16.2, text p. 745). (b) The chemical shift of the benzene protons is at considerably greater chemical shift because benzene is aromatic and 1,4-cyclohexadiene is not. 16.6 (b) Among other features, the NMR spectrum of 1-bromo-4-ethylbenzene has a typical ethyl quartet and a typical para-substitution pattern for the ring protons, as shown in Fig. 16.3, text p. 747, whereas the spectrum of (2- bromoethyl)benzene should show a pair of triplets for the methylene protons and a complex pattern for the ring protons. If this isn’t enough to distinguish the two compounds, the integral of the ring protons relative to the integral of the remaining protons is different in the two compounds. 16.7 (b) The IR spectrum indicates the presence of an OH group, and the chemical shift of the broad NMR resonance (d 6.0) suggests that this could be a phenol. The splitting patterns of the d 1.17 and d 2.58 resonances show that the compound also contains an ethyl group, and the splitting pattern of the ring protons shows that the compound is a para-disubstituted benzene derivative. -

Docw R S Acquired by ERIC Include Many Informal Unpublished * Material - T Available Fromother Sources
DOCUMENT RESUME ED 135 584 SE 021 575 AUTHOR Zdravkovich, V. TITLE Organic Chemistry Self Instructional Package 4: Alkanes-Nomenclature. INSTITUTION Prince George's Community Coll., Largo, Md. PUB DATE 76 NOTE 41p.; For related Packages 1-17, see SE 021 572-588; Not available in hard copy due to copyright restrictions AVAILABLE FROM Prince George's Community College Bookstore, Largo, Maryland 20870 ($17.00 a set, $1.00 each) EDRS PRICE MF-$0.83 Plus Postage. MC Not Available from EDRS. DESCRIPTORS *Autoinstructional Aids; *Chemistry; *College Science; Higher Education; *Independent Study; Individualized Instruction; Individualized Programs; *Organic Chemistry; Science Education; Self Help Programs IDENTIFIERS Alkanes; Prince Georges Community College ABSTRACT This booklet, one of a series of 17 developed at Prince George's Community College, largo, Maryland, provides an individualized, self-paced undergraduate organic chemistry instruction module designed to augment any course in organic chemistry but particularly those taught using the text "Organic Chemistry" by Morrison and Boyd. The entire series of modules covers the first 13 chapters of the Morrison-Boyd text in great detail. Each module has been provided with from one to three audiotapes, available from PrinOe George's Community College, to provide students additional explanations of particular concepts. Each module includes a self-evaluation exercise, a reference guide, worksheets to be completed with the audiotapes, answer sheets for the worksheets, a progress evaluation, an answer sheet for the progress evaluation, an answer sheet for the self-evaluation exercise, an introduction to the topic covered by the module, and student performance objectives for the module. The topic of this module is alkanes. -

Organic and Biological Chemistry
CHAPTER 23 Organic and Biological Chemistry CONTENTS HO H 23.1 ▶ Organic Molecules and Their C Structures: Alkanes H H C 23.2 ▶ Families of Organic Compounds: HO O O C C Functional Groups 23.3 ▶ Naming Organic Compounds H CC 23.4 ▶ Carbohydrates:HO A Biological Example HO OH of Isomers H 23.5 ▶ Valence Bond TCheory and Orbital OverlapH Pictures H C 23.6 ▶ Lipids:HO A Biological EOxample ofO Cis–Trans IsomerismC C 23.7 ▶ Formal Charge and Resonance in Organic CompoundsH CC 23.8 ▶ Conjugated SystemsHO OH 23.9 ▶ Proteins: A Biological Example of Conjugation 23.10 ▶ Aromatic Compounds and Molecular Ascorbic acid, also known as vitamin C, is an essential nutrient in the human diet Orbital Theory because it is not synthesized in our body. We can eat citrus fruits or take pills that contain vitamin C to maintain health. 23.11 ▶ Nucleic Acids: A Biological Example of Aromaticity ? Which Is Better, Natural or Synthetic? The answer to this question can be found in the INQUIRY ▶▶▶ on page 1021. STUDY GUIDE M23_MCMU3170_07_SE_C23.indd 978 27/11/14 2:11 AM f the ultimate goal of chemistry is to understand the world around us on a molecular level, then a knowledge of biochemistry—the chemistry of living organisms—is a central part Iof that goal. Biochemistry, in turn, is a branch of organic chemistry, a term originally used to mean the study of compounds from living organisms while inorganic chemistry was used for the study of compounds from nonliving sources. Today, however, we know that there are no fundamental differences between organic and inorganic compounds; the same principles apply to both. -

Synthesis of Novel Extremely Sterically Hindered Tertiary Alkylamines
Synthesis of Novel Extremely Sterically Hindered Tertiary Alkylamines von der Fakultät für Naturwissenschaften der Technischen Universität Chemnitz genehmigte Dissertation zur Erlangung des akademischen Grades doktor rerum naturalium (Dr. rer. nat.) vorgelegt von M.Sc. Tharallah A. Shoker geboren am 18. April 1985 in Nabi Chit, Lebanon eingereicht am 17. January 2018 Gutachter: Prof. Dr. Klaus Banert JP Dr. Evgeny Kataev Tag der Verteidigung: 16. April 2018 Die vorliegende Arbeit wurde in der Zeit von Oktober 2015 bis Oktober 2017 am Institut für Chemie an der Fakultät für Naturwissenschaften der Technischen Universität Chemnitz unter der Leitung von Prof. Dr. Klaus Banert angefertigt. 2 Bibliographic Description Synthesis of Novel Extremely Sterically Hindered Tertiary Alkylamines SHOKER, THARALLAH Technische Universität Chemnitz, Fakultät für Naturwissenschaften Dissertation, 2017, 208 pages. Abstract Three advanced methodologies for the preparation of extremely sterically hindered tertiary alkyl amines have been developed. The syntheses of 28 novel tertiary alkylamines that accommodate unusual steric hindrance are detailed. The electrophilic amination of alkyl Grignard reagents with N-chlorodialkylamines, in the presence of N,N,N′,N′-tetramethylethylenediamine (TMEDA) as a key additive, gives a variety of unprecedentedly sterically hindered tertiary alkylamines in good yields. Alternative strategy to 1-adamantyl-substituted (1-Ad) sterically hindered tertiary amines, which involved instead an SN1 reaction between 1-Ad cation with various secondary amines, is described. A complementary strategy to 1-Ad-based sterically hindered tertiary amines, which involves an iminium salt intermediate, is also reported. Salient features of the three protocols that are detailed here include unusual tolerance of steric hindrance, mild reaction conditions employed, ease of product isolation-purification, and absence of catalysts/transition metals. -
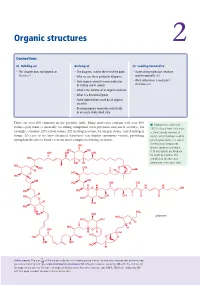
Organic Structures 2
Organic structures 2 Connections Building on Arriving at Looking forward to • This chapter does not depend on • The diagrams used in the rest of the book • Ascertaining molecular structure Chapter 1 • Why we use these particular diagrams spectroscopically ch3 • How organic chemists name molecules • What determines a molecule’s in writing and in speech structure ch4 • What is the skeleton of an organic molecule • What is a functional group • Some abbreviations used by all organic chemists • Drawing organic molecules realistically in an easily understood style There are over 100 elements in the periodic table. Many molecules contain well over 100 ■ Palytoxin was isolated in atoms—palytoxin (a naturally occurring compound with potential anticancer activity), for 1971 in Hawaii from Limu make example, contains 129 carbon atoms, 221 hydrogen atoms, 54 oxygen atoms, and 3 nitrogen o Hane (‘deadly seaweed of atoms. It’s easy to see how chemical structures can display enormous variety, providing Hana’), which had been used to enough molecules to build even the most complicated living creatures. poison spear points. It is one of the most toxic compounds OH known, requiring only about HO OH 0.15 micrograms per kilogram OH OH OH for death by injection. The HO complicated structure was O OH H determined a few years later. HO OH OH O H H O O HO HO H OH OH HO OH OH H H H H O N N OH HO HO H HO OH OH O O O HO OH H OH HO OH NH2 HO OH HO palytoxin HO OH O H HO H OH O H H H HO HO OH O O OH OH HO H Online support. -

Approach and Synthesis of Strychnos Alkaloids
N° d'ordre : 4155 THÈSE Présentée à L'UNIVERSITÉ BORDEAUX I ÉCOLE DOCTORALE DES SCIENCES CHIMIQUES par Dawood Hosni DAWOOD POUR OBTENIR LE GRADE DE DOCTEUR SPÉCIALITÉ : CHIMIE ORGANIQUE ********************* TOWARDS THE SYNTHESIS OF MONOTERPENOIDS INDOLE ALKALOIDS OF THE ASPIDOSPERMATAN AND STRYCHNAN TYPE ********************* Soutenue le: 17 décembre 2010 Après avis de: MM. PIVA Olivier Professeur, Claude Bernard Lyon 1 Rapporteur PALE Patrick Professeur, Louis Pasteur Strasbourg 1 Rapporteur Devant la commission d'examen formée de : MM. PIVA Olivier Professeur, Claude Bernard Lyon 1 Rapporteur PALE Patrick Professeur, Louis Pasteur Strasbourg 1 Rapporteur POISSON Jean-François Chargé de recherche, CNRS Examinateur VINCENT Jean-Marc Directeur de recherche, CNRS Examinateur LANDAIS Yannick Professeur, Bordeaux 1 Directeur de thèse ROBERT Frédéric Chargé de recherche, CNRS Codirecteur de thèse - 2010 - Abbreviations ∆: reflux °C: celsius degrees Ac: acetyle ALB Aluminium Lithium bis(binaphthoxide) complex AIBN : azobis(isobutyronitrile) aq.: aqueous Ar : aromatic BINAP : 2,2'-bis(diphenylphosphino)-1,1'-binaphthyle BINAPO : 2-diphenylphosphino-2'-diphenylphosphinyl-1,1'-binaphthalene BINOL: 1,1’-bi-2-naphthol Boc: tert-butyloxycarbonyle BOX: Bisoxazoline Bz : benzoyle Bn: benzyle cat. : catalytic DBU: 1,8-diazabicyclo[5.4.0]undec-7-ene DCM: dichloromethane DCC: dicyclohexacarbodiimide dr.: diastereomeric ratio DIBAL-H: diisobutylaluminium hydride DIPEA: diisopropyléthylamine (Hünig Base) DMAP: dimethylaminopyridine DME: dimethoxyethane -

United States Patent Office Patiented Jan
3,299,012 United States Patent Office Patiented Jan. 17, 1967 2 methacrylates, vinyl chloride, vinylidine chloride, vinyli 3,299,92 dine fluoride, vinyl acetate, ethylene, propylene, isobutyl PROCESS FOR THE PREPARATION OF ene, O-methylstyrene, and styrene. POLYLACTONES Rciand 5. Kern, Bridgeton, Mo., assignor to Monsanto According to the instant invention, a rearrangement re Conapany, a corporation of Delaware action, catalyzed by a Lewis acid, is used to prepare No Drawing. Filled Dec. 17, 1963, Ser. No. 331,096 polymeric substances characterized by lactone links and 12 Claims. (C. 269-78.5) also carboxylic acid groups or carboxylic acid ester groups along the polymer chain. This invention relates to a process for the preparation It will be understood that various vinyl ethers, as de of polylactones, and to ester derivatives of polylactones. 10 scribed above, can be used to prepare the polymeric re This invention particularly relates to lactones derived actant, however for convenience the invention is de from alternating copolymers of certain vinyl ethers and Scribed with reference to the tert-butyl vinyl ether co unsaturated carboxylic acid anhydrides. polymer. The process can be practiced when the group It is an object of this invention to provide a novel attached to the vinyl group through an ether link is one process for preparing polylactones. 5 known to form a stable carbonium ion. It is another object of this invention to provide a simple The rearrangement of the vinyl ether/olefinic carboxylic direct method for converting vinyl ether/unsaturated car anhydride copolymers is conducted in the presence of any boxylic acid anhydride copolymers to polymers charac of the materials known as acids within the concept of terized by lactone rings along the polymer chains, and Dr. -

Total Synthesis of Ancistrotanzanine A
TOTAL SYNTHESIS OF ANCISTROTANZANINE A A Thesis Submitted in Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Chemistry at The University of Adelaide School of Chemistry and Physics By Jason Stewart Brusnahan B. Sc. (Honours) October 2009 ‘If it’s not red, it’s dead.’ Jason Brusnahan i Table of Contents Chapter 1: Introduction 1.1 General Introduction 2 1.2 The Atropisomerism Phenomenon 7 1.3 Total Synthesis 10 1.3.1 Intramolecular Approach 10 1.3.2 Intermolecular Approach 14 1.3.2.1 Application of the Meyers Biaryl Synthesis 14 1.3.2.2 Cross Coupling 19 1.4 Sterically Challenging Naphthylisoquinoline Alkaloids 26 1.5 Aryllead Triacetates 28 1.6 Application of the Pinhey-Barton Reaction to the Total Synthesis of Ancistrocladidine 31 1.7 Work Described in this Thesis 38 1.8 References 41 Chapter 2: Studies into a Diastereoselective Pinhey-Barton Reaction 2.1 Application of the Pinhey-Barton Reaction to the Total Synthesis of Ancistrotanzanine A 46 2.2 Search for a chiral ligand 47 2.3 Combing the ‘Lactone Method’ with the Pinhey-Barton Reaction 53 2.4 Preparation of the Aryllead Species 2.1 55 2.5 Formation of the 5,3-Biaryl Bond 57 2.6 Investigations into a Diastereoselective Pinhey-Barton Reaction 60 ii 2.7 Conclusions 66 2.8 References 68 Chapter 3: The Total Synthesis of Ancistrotanzanine A 3.1 Strategies Utilised in the Synthesis of 3,4-Dihydroisoquinolines 70 3.1.1 Introduction of the Nitrogen Functionality via the Henry Reaction 71 3.1.2 Introduction of Chirality via the Reaction of Grignard Reagents with -

The Institute of Paper Chemistry
The Institute of Paper Chemistry Appleton, Wisconsin Doctor's Dissertation Anhydride Derivatives of Trimellitic Anhydride Richard G. Barker June, 1963 ANHYDRIDE DERIVATIVES OF TRIMELLITIC ANHYDRIDE A thesis submitted by Richard G. Barker B.A. 1958, Hamilton College M.S. 1960, Lawrence College in partial fulfillment of the requirements of The Institute of Paper Chemistry for the degree of Doctor of Philosophy from Lawrence College Appleton, Wisconsin June, 1963 TABLE OF CONTENTS Page GENERAL SUMMARY 1 INTRODUCTION 3 Trimellitic Anhydride 3 Unsymmetrical Anhydrides 5 Preparation of Unsymmetrical Anhydrides 6 Disproportionation of Unsymmetrical Anhydrides 8 Hydrolysis of Unsymmetrical Anhydrides 11 Reactions of Unsymmetrical Anhydrides 12 Previous Investigations on the Esterification of Unsymmetrical Anhydrides 12 The Mechanism of Acylation by Unsymmetrical Anhydrides 13 Trifluoroacetic Anhydride as an Esterification Promoter 16 PRESENTATION AND APPROACH TO THE PROBLEM 22 PROCEDURES AND RESULTS 24 The Trimellitic Anhydride-Trifluoroacetic Anhydride Reaction System 24 Preparation of the Unsymmetrical Anhydride of TMA and TFAA 24 Purification and Analysis of the Unsymmetrical Anhydride of TMA and TFAA 25 Disproportionation of the Unsymmetrical Anhydride of TMA and TFAA 27 Hydrolysis of the Unsymmetrical Anhydride of TMA and TFAA 28 Esterification of the Unsymmetrical Anhydride of TMA and TFAA 29 The Unsymmetrical Anhydride of Trimellitic and Acetic Anhydrides 31 Preparation of the Unsymmetrical Anhydride 31 Purification and Analysis of the -

“Butyl” and “Pentyl”
1/31/18 CHEM 109A Organic Chemistry https://labs.chem.ucsb.edu/zakarian/armen/courses.html Chapter 3 An Introduction to Organic Compounds Nomenclature, Physical Properties, and Structure © 2017 Pearson Education, Inc. How many hydrogens are attached to each carbon HO cholesterol © 2017 Pearson Education, Inc. 1 1/31/18 Alkanes are Saturated Hydrocarbons, CnH2n+2, Only Single Bonds heating power bbq lighters gasoline jet fuel diesel © 2017 Pearson Education, Inc. Types of Alkanes in Chapter 3 straight chain branched cyclic (CnH2n+2) (C H ) n 2n+2 (CnH2n) hexane cyclohexane butane © 2017 Pearson Education, Inc. 2 1/31/18 Alkanes and Alkyl Groups: Two alkanes C4H10 CH3CH2CH2CH2−X X Butane Butyl group and X X CH3CH2CH2CH3 CH3CH2CHCH2 Butane sec-butyl group and Isobutane X CH3CH(CH3)2 X and CH3 (CH3)3C−X CH3 CH (CH3)2CHCH2−X CH3 Isobutane isobutyl group tert-butyl group © 2017 Pearsonbutane Education, Inc. and isobutane are constitutional isomers Alkyl Substituents X X X X ethyl group n-propyl group n-butyl group n-pentyl group R X generic alkyl group © 2017 Pearson Education, Inc. 3 1/31/18 n = an Unbranched Chain Common names sometimes use “n” (stands for “normal”) to indicate a straight-chain alkane. © 2017 Pearson Education, Inc. Alkyl Substituents X X X X ethyl group n-propyl group n-butyl group n-pentyl group X R X X sec-butyl group isobutyl group generic alkyl group branched alkyl groups © 2017 Pearson Education, Inc. 4 1/31/18 Primary, Secondary, and Tertiary Carbons • A primary carbon is bonded to one carbon.