An Analysis of Ancestral Sequence Resurrection in the Context of Guanylate Kinase Evolution
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Non-Homologous Isofunctional Enzymes: a Systematic Analysis Of
Omelchenko et al. Biology Direct 2010, 5:31 http://www.biology-direct.com/content/5/1/31 RESEARCH Open Access Non-homologousResearch isofunctional enzymes: A systematic analysis of alternative solutions in enzyme evolution Marina V Omelchenko, Michael Y Galperin*, Yuri I Wolf and Eugene V Koonin Abstract Background: Evolutionarily unrelated proteins that catalyze the same biochemical reactions are often referred to as analogous - as opposed to homologous - enzymes. The existence of numerous alternative, non-homologous enzyme isoforms presents an interesting evolutionary problem; it also complicates genome-based reconstruction of the metabolic pathways in a variety of organisms. In 1998, a systematic search for analogous enzymes resulted in the identification of 105 Enzyme Commission (EC) numbers that included two or more proteins without detectable sequence similarity to each other, including 34 EC nodes where proteins were known (or predicted) to have distinct structural folds, indicating independent evolutionary origins. In the past 12 years, many putative non-homologous isofunctional enzymes were identified in newly sequenced genomes. In addition, efforts in structural genomics resulted in a vastly improved structural coverage of proteomes, providing for definitive assessment of (non)homologous relationships between proteins. Results: We report the results of a comprehensive search for non-homologous isofunctional enzymes (NISE) that yielded 185 EC nodes with two or more experimentally characterized - or predicted - structurally unrelated proteins. Of these NISE sets, only 74 were from the original 1998 list. Structural assignments of the NISE show over-representation of proteins with the TIM barrel fold and the nucleotide-binding Rossmann fold. From the functional perspective, the set of NISE is enriched in hydrolases, particularly carbohydrate hydrolases, and in enzymes involved in defense against oxidative stress. -

Guanylate Kinase (Ec 2.7.4.8)
Enzymatic Assay of GUANYLATE KINASE (EC 2.7.4.8) PRINCIPLE: Guanylate Kinase GMP + ATP > GDP + ADP Pyruvate Kinase ADP + PEP > ATP + Pyruvate Pyruvate Kinase GDP + PEP > GTP + Pyruvate Lactic Dehydrogenase 2 Pyruvate + 2 ß-NADH > 2 Lactate + 2 ß-NAD Abbreviations used: GMP = Guanosine 5'-Monophosphate ATP = Adenosine 5'-Triphosphate GDP = Guanosine 5'-Diphosphate ADP = Adenosine 5'-Diphosphate PEP = Phospho(enol)phosphate ß-NADH = ß-Nicotinamide Adenine Dinucleotide, Reduced Form ß-NAD = ß-Nicotinamide Adenine Dinucleotide, Oxidized Form CONDITIONS: T = 30°C, pH = 7.5, A340nm, Light path = 1 cm METHOD: Continuous Spectrophotometric Rate Determination REAGENTS: A. 200 mM Tris HCl Buffer, pH 7.5 at 30°C (Prepare 50 ml in deionized water using Trizma Base, Sigma Prod. No. T-1503. Adjust to pH 7.5 at 30°C with 1 M HCl.) B. 1 M Potassium Chloride Solution (KCl) (Prepare 10 ml in deionized water using Potassium Chloride, Sigma Prod. No. P-4504.) C. 60 mM Magnesium Sulfate Solution (MgSO4) (Prepare 20 ml in deionized water using Magnesium Sulfate, Heptahydrate, Sigma Prod. No. M-1880.) D. 40 mM Phospho(enol)pyruvate Solution (PEP) (Prepare 50 ml in deionized water using Phospho(enol)Pyruvate, Trisodium Salt, Hydrate, Sigma Prod. No. P-7002. PREPARE FRESH.) Revised: 03/10/94 Page 1 of 4 Enzymatic Assay of GUANYLATE KINASE (EC 2.7.4.8) REAGENTS: (continued) E. 100 mM Ethylenediaminetetraacetic Acid Solution (EDTA) (Prepare 10 ml in deionized water using Ethylenediaminetetraacetic Acid, Tetrasodium Salt, Hydrate, Sigma Stock No. ED4SS.) F. 3.8 mM ß-Nicotinamide Adenine Dinucleotide, Reduced Form (ß-NADH) (Prepare 2 ml in deionized water using ß-Nicotinamide Adenine Dinucleotide, Reduced Form, Dipotassium Salt, Sigma Prod. -

WO 2013/180584 Al 5 December 2013 (05.12.2013) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2013/180584 Al 5 December 2013 (05.12.2013) P O P C T (51) International Patent Classification: AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, C12N 1/21 (2006.01) C12N 15/74 (2006.01) BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, C12N 15/52 (2006.01) C12P 5/02 (2006.01) DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, C12N 15/63 (2006.01) HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KN, KP, KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, (21) International Application Number: MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, PCT/NZ20 13/000095 OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SC, (22) International Filing Date: SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, 4 June 2013 (04.06.2013) TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (26) Publication Language: English GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, SZ, TZ, (30) Priority Data: UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, 61/654,412 1 June 2012 (01 .06.2012) US TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, LV, (71) Applicant: LANZATECH NEW ZEALAND LIMITED MC, MK, MT, NL, NO, PL, PT, RO, RS, SE, SI, SK, SM, [NZ/NZ]; 24 Balfour Road, Parnell, Auckland, 1052 (NZ). -

(12) Patent Application Publication (10) Pub. No.: US 2014/0155567 A1 Burk Et Al
US 2014O155567A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2014/0155567 A1 Burk et al. (43) Pub. Date: Jun. 5, 2014 (54) MICROORGANISMS AND METHODS FOR (60) Provisional application No. 61/331,812, filed on May THE BIOSYNTHESIS OF BUTADENE 5, 2010. (71) Applicant: Genomatica, Inc., San Diego, CA (US) Publication Classification (72) Inventors: Mark J. Burk, San Diego, CA (US); (51) Int. Cl. Anthony P. Burgard, Bellefonte, PA CI2P 5/02 (2006.01) (US); Jun Sun, San Diego, CA (US); CSF 36/06 (2006.01) Robin E. Osterhout, San Diego, CA CD7C II/6 (2006.01) (US); Priti Pharkya, San Diego, CA (52) U.S. Cl. (US) CPC ................. CI2P5/026 (2013.01); C07C II/I6 (2013.01); C08F 136/06 (2013.01) (73) Assignee: Genomatica, Inc., San Diego, CA (US) USPC ... 526/335; 435/252.3:435/167; 435/254.2: (21) Appl. No.: 14/059,131 435/254.11: 435/252.33: 435/254.21:585/16 (22) Filed: Oct. 21, 2013 (57) ABSTRACT O O The invention provides non-naturally occurring microbial Related U.S. Application Data organisms having a butadiene pathway. The invention addi (63) Continuation of application No. 13/101,046, filed on tionally provides methods of using Such organisms to produce May 4, 2011, now Pat. No. 8,580,543. butadiene. Patent Application Publication Jun. 5, 2014 Sheet 1 of 4 US 2014/O155567 A1 ?ueudos!SMS |?un61– Patent Application Publication Jun. 5, 2014 Sheet 2 of 4 US 2014/O155567 A1 VOJ OO O Z?un61– Patent Application Publication US 2014/O155567 A1 {}}} Hººso Patent Application Publication Jun. -

Table S1. List of Oligonucleotide Primers Used
Table S1. List of oligonucleotide primers used. Cla4 LF-5' GTAGGATCCGCTCTGTCAAGCCTCCGACC M629Arev CCTCCCTCCATGTACTCcgcGATGACCCAgAGCTCGTTG M629Afwd CAACGAGCTcTGGGTCATCgcgGAGTACATGGAGGGAGG LF-3' GTAGGCCATCTAGGCCGCAATCTCGTCAAGTAAAGTCG RF-5' GTAGGCCTGAGTGGCCCGAGATTGCAACGTGTAACC RF-3' GTAGGATCCCGTACGCTGCGATCGCTTGC Ukc1 LF-5' GCAATATTATGTCTACTTTGAGCG M398Arev CCGCCGGGCAAgAAtTCcgcGAGAAGGTACAGATACGc M398Afwd gCGTATCTGTACCTTCTCgcgGAaTTcTTGCCCGGCGG LF-3' GAGGCCATCTAGGCCATTTACGATGGCAGACAAAGG RF-5' GTGGCCTGAGTGGCCATTGGTTTGGGCGAATGGC RF-3' GCAATATTCGTACGTCAACAGCGCG Nrc2 LF-5' GCAATATTTCGAAAAGGGTCGTTCC M454Grev GCCACCCATGCAGTAcTCgccGCAGAGGTAGAGGTAATC M454Gfwd GATTACCTCTACCTCTGCggcGAgTACTGCATGGGTGGC LF-3' GAGGCCATCTAGGCCGACGAGTGAAGCTTTCGAGCG RF-5' GAGGCCTGAGTGGCCTAAGCATCTTGGCTTCTGC RF-3' GCAATATTCGGTCAACGCTTTTCAGATACC Ipl1 LF-5' GTCAATATTCTACTTTGTGAAGACGCTGC M629Arev GCTCCCCACGACCAGCgAATTCGATagcGAGGAAGACTCGGCCCTCATC M629Afwd GATGAGGGCCGAGTCTTCCTCgctATCGAATTcGCTGGTCGTGGGGAGC LF-3' TGAGGCCATCTAGGCCGGTGCCTTAGATTCCGTATAGC RF-5' CATGGCCTGAGTGGCCGATTCTTCTTCTGTCATCGAC RF-3' GACAATATTGCTGACCTTGTCTACTTGG Ire1 LF-5' GCAATATTAAAGCACAACTCAACGC D1014Arev CCGTAGCCAAGCACCTCGgCCGAtATcGTGAGCGAAG D1014Afwd CTTCGCTCACgATaTCGGcCGAGGTGCTTGGCTACGG LF-3' GAGGCCATCTAGGCCAACTGGGCAAAGGAGATGGA RF-5' GAGGCCTGAGTGGCCGTGCGCCTGTGTATCTCTTTG RF-3' GCAATATTGGCCATCTGAGGGCTGAC Kin28 LF-5' GACAATATTCATCTTTCACCCTTCCAAAG L94Arev TGATGAGTGCTTCTAGATTGGTGTCggcGAAcTCgAGCACCAGGTTG L94Afwd CAACCTGGTGCTcGAgTTCgccGACACCAATCTAGAAGCACTCATCA LF-3' TGAGGCCATCTAGGCCCACAGAGATCCGCTTTAATGC RF-5' CATGGCCTGAGTGGCCAGGGCTAGTACGACCTCG -

A Global Analysis of Enzyme Compartmentalization to Glycosomes
pathogens Article A Global Analysis of Enzyme Compartmentalization to Glycosomes Hina Durrani 1, Marshall Hampton 2 , Jon N. Rumbley 3 and Sara L. Zimmer 1,* 1 Department of Biomedical Sciences, University of Minnesota Medical School, Duluth Campus, Duluth, MN 55812, USA; [email protected] 2 Mathematics & Statistics Department, University of Minnesota Duluth, Duluth, MN 55812, USA; [email protected] 3 College of Pharmacy, University of Minnesota, Duluth Campus, Duluth, MN 55812, USA; [email protected] * Correspondence: [email protected] Received: 25 March 2020; Accepted: 9 April 2020; Published: 12 April 2020 Abstract: In kinetoplastids, the first seven steps of glycolysis are compartmentalized into a glycosome along with parts of other metabolic pathways. This organelle shares a common ancestor with the better-understood eukaryotic peroxisome. Much of our understanding of the emergence, evolution, and maintenance of glycosomes is limited to explorations of the dixenous parasites, including the enzymatic contents of the organelle. Our objective was to determine the extent that we could leverage existing studies in model kinetoplastids to determine the composition of glycosomes in species lacking evidence of experimental localization. These include diverse monoxenous species and dixenous species with very different hosts. For many of these, genome or transcriptome sequences are available. Our approach initiated with a meta-analysis of existing studies to generate a subset of enzymes with highest evidence of glycosome localization. From this dataset we extracted the best possible glycosome signal peptide identification scheme for in silico identification of glycosomal proteins from any kinetoplastid species. Validation suggested that a high glycosome localization score from our algorithm would be indicative of a glycosomal protein. -

The Metabolic Building Blocks of a Minimal Cell Supplementary
The metabolic building blocks of a minimal cell Mariana Reyes-Prieto, Rosario Gil, Mercè Llabrés, Pere Palmer and Andrés Moya Supplementary material. Table S1. List of enzymes and reactions modified from Gabaldon et. al. (2007). n.i.: non identified. E.C. Name Reaction Gil et. al. 2004 Glass et. al. 2006 number 2.7.1.69 phosphotransferase system glc + pep → g6p + pyr PTS MG041, 069, 429 5.3.1.9 glucose-6-phosphate isomerase g6p ↔ f6p PGI MG111 2.7.1.11 6-phosphofructokinase f6p + atp → fbp + adp PFK MG215 4.1.2.13 fructose-1,6-bisphosphate aldolase fbp ↔ gdp + dhp FBA MG023 5.3.1.1 triose-phosphate isomerase gdp ↔ dhp TPI MG431 glyceraldehyde-3-phosphate gdp + nad + p ↔ bpg + 1.2.1.12 GAP MG301 dehydrogenase nadh 2.7.2.3 phosphoglycerate kinase bpg + adp ↔ 3pg + atp PGK MG300 5.4.2.1 phosphoglycerate mutase 3pg ↔ 2pg GPM MG430 4.2.1.11 enolase 2pg ↔ pep ENO MG407 2.7.1.40 pyruvate kinase pep + adp → pyr + atp PYK MG216 1.1.1.27 lactate dehydrogenase pyr + nadh ↔ lac + nad LDH MG460 1.1.1.94 sn-glycerol-3-phosphate dehydrogenase dhp + nadh → g3p + nad GPS n.i. 2.3.1.15 sn-glycerol-3-phosphate acyltransferase g3p + pal → mag PLSb n.i. 2.3.1.51 1-acyl-sn-glycerol-3-phosphate mag + pal → dag PLSc MG212 acyltransferase 2.7.7.41 phosphatidate cytidyltransferase dag + ctp → cdp-dag + pp CDS MG437 cdp-dag + ser → pser + 2.7.8.8 phosphatidylserine synthase PSS n.i. cmp 4.1.1.65 phosphatidylserine decarboxylase pser → peta PSD n.i. -
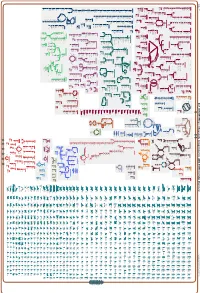
Generate Metabolic Map Poster
Authors: Pallavi Subhraveti Ron Caspi Peter Midford Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_003855395Cyc: Shewanella livingstonensis LMG 19866 Cellular Overview Connections between pathways are omitted for legibility. -

Supplementary Informations SI2. Supplementary Table 1
Supplementary Informations SI2. Supplementary Table 1. M9, soil, and rhizosphere media composition. LB in Compound Name Exchange Reaction LB in soil LBin M9 rhizosphere H2O EX_cpd00001_e0 -15 -15 -10 O2 EX_cpd00007_e0 -15 -15 -10 Phosphate EX_cpd00009_e0 -15 -15 -10 CO2 EX_cpd00011_e0 -15 -15 0 Ammonia EX_cpd00013_e0 -7.5 -7.5 -10 L-glutamate EX_cpd00023_e0 0 -0.0283302 0 D-glucose EX_cpd00027_e0 -0.61972444 -0.04098397 0 Mn2 EX_cpd00030_e0 -15 -15 -10 Glycine EX_cpd00033_e0 -0.0068175 -0.00693094 0 Zn2 EX_cpd00034_e0 -15 -15 -10 L-alanine EX_cpd00035_e0 -0.02780553 -0.00823049 0 Succinate EX_cpd00036_e0 -0.0056245 -0.12240603 0 L-lysine EX_cpd00039_e0 0 -10 0 L-aspartate EX_cpd00041_e0 0 -0.03205557 0 Sulfate EX_cpd00048_e0 -15 -15 -10 L-arginine EX_cpd00051_e0 -0.0068175 -0.00948672 0 L-serine EX_cpd00054_e0 0 -0.01004986 0 Cu2+ EX_cpd00058_e0 -15 -15 -10 Ca2+ EX_cpd00063_e0 -15 -100 -10 L-ornithine EX_cpd00064_e0 -0.0068175 -0.00831712 0 H+ EX_cpd00067_e0 -15 -15 -10 L-tyrosine EX_cpd00069_e0 -0.0068175 -0.00233919 0 Sucrose EX_cpd00076_e0 0 -0.02049199 0 L-cysteine EX_cpd00084_e0 -0.0068175 0 0 Cl- EX_cpd00099_e0 -15 -15 -10 Glycerol EX_cpd00100_e0 0 0 -10 Biotin EX_cpd00104_e0 -15 -15 0 D-ribose EX_cpd00105_e0 -0.01862144 0 0 L-leucine EX_cpd00107_e0 -0.03596182 -0.00303228 0 D-galactose EX_cpd00108_e0 -0.25290619 -0.18317325 0 L-histidine EX_cpd00119_e0 -0.0068175 -0.00506825 0 L-proline EX_cpd00129_e0 -0.01102953 0 0 L-malate EX_cpd00130_e0 -0.03649016 -0.79413596 0 D-mannose EX_cpd00138_e0 -0.2540567 -0.05436649 0 Co2 EX_cpd00149_e0 -

Spatial and Temporal Aspects of Signalling 6 1
r r r Cell Signalling Biology Michael J. Berridge Module 6 Spatial and Temporal Aspects of Signalling 6 1 Module 6 Spatial and Temporal Aspects of Signalling Synopsis The function and efficiency of cell signalling pathways are very dependent on their organization both in space and time. With regard to spatial organization, signalling components are highly organized with respect to their cellular location and how they transmit information from one region of the cell to another. This spatial organization of signalling pathways depends on the molecular interactions that occur between signalling components that use signal transduction domains to construct signalling pathways. Very often, the components responsible for information transfer mechanisms are held in place by being attached to scaffolding proteins to form macromolecular signalling complexes. Sometimes these macromolecular complexes can be organized further by being localized to specific regions of the cell, as found in lipid rafts and caveolae or in the T-tubule regions of skeletal and cardiac cells. Another feature of the spatial aspects concerns the local Another important temporal aspect is timing and signal and global aspects of signalling. The spatial organization of integration, which relates to the way in which functional signalling molecules mentioned above can lead to highly interactions between signalling pathways are determined localized signalling events, but when the signalling mo- by both the order and the timing of their presentations. lecules are more evenly distributed, signals can spread The organization of signalling systems in both time and more globally throughout the cell. In addition, signals space greatly enhances both their efficiency and versatility. -

Structure of Human Adenosine Kinase at 1.5 Å Resolution†,‡ Irimpan I
Biochemistry 1998, 37, 15607-15620 15607 Structure of Human Adenosine Kinase at 1.5 Å Resolution†,‡ Irimpan I. Mathews,§ Mark D. Erion,| and Steven E. Ealick*,§ Department of Chemistry and Chemical Biology, Cornell UniVersity, Ithaca, New York 14853, and Metabasis Therapeutics, Inc., 9390 Town Center DriVe, San Diego, California 92121 ReceiVed June 29, 1998; ReVised Manuscript ReceiVed September 9, 1998 ABSTRACT: Adenosine kinase (AK) is a key enzyme in the regulation of extracellular adenosine and intracellular adenylate levels. Inhibitors of adenosine kinase elevate adenosine to levels that activate nearby adenosine receptors and produce a wide variety of therapeutically beneficial activities. Accordingly, AK is a promising target for new analgesic, neuroprotective, and cardioprotective agents. We determined the structure of human adenosine kinase by X-ray crystallography using MAD phasing techniques and refined the structure to 1.5 Å resolution. The enzyme structure consisted of one large R/â domain with nine â-strands, eight R-helices, and one small R/â-domain with five â-strands and two R-helices. The active site is formed along the edge of the â-sheet in the large domain while the small domain acts as a lid to cover the upper face of the active site. The overall structure is similar to the recently reported structure of ribokinase from Escherichia coli [Sigrell et al. (1998) Structure 6, 183-193]. The structure of ribokinase was determined at 1.8 Å resolution and represents the first structure of a new family of carbohydrate kinases. Two molecules of adenosine were present in the AK crystal structure with one adenosine molecule located in a site that matches the ribose site in ribokinase and probably represents the substrate-binding site. -

O O2 Enzymes Available from Sigma Enzymes Available from Sigma
COO 2.7.1.15 Ribokinase OXIDOREDUCTASES CONH2 COO 2.7.1.16 Ribulokinase 1.1.1.1 Alcohol dehydrogenase BLOOD GROUP + O O + O O 1.1.1.3 Homoserine dehydrogenase HYALURONIC ACID DERMATAN ALGINATES O-ANTIGENS STARCH GLYCOGEN CH COO N COO 2.7.1.17 Xylulokinase P GLYCOPROTEINS SUBSTANCES 2 OH N + COO 1.1.1.8 Glycerol-3-phosphate dehydrogenase Ribose -O - P - O - P - O- Adenosine(P) Ribose - O - P - O - P - O -Adenosine NICOTINATE 2.7.1.19 Phosphoribulokinase GANGLIOSIDES PEPTIDO- CH OH CH OH N 1 + COO 1.1.1.9 D-Xylulose reductase 2 2 NH .2.1 2.7.1.24 Dephospho-CoA kinase O CHITIN CHONDROITIN PECTIN INULIN CELLULOSE O O NH O O O O Ribose- P 2.4 N N RP 1.1.1.10 l-Xylulose reductase MUCINS GLYCAN 6.3.5.1 2.7.7.18 2.7.1.25 Adenylylsulfate kinase CH2OH HO Indoleacetate Indoxyl + 1.1.1.14 l-Iditol dehydrogenase L O O O Desamino-NAD Nicotinate- Quinolinate- A 2.7.1.28 Triokinase O O 1.1.1.132 HO (Auxin) NAD(P) 6.3.1.5 2.4.2.19 1.1.1.19 Glucuronate reductase CHOH - 2.4.1.68 CH3 OH OH OH nucleotide 2.7.1.30 Glycerol kinase Y - COO nucleotide 2.7.1.31 Glycerate kinase 1.1.1.21 Aldehyde reductase AcNH CHOH COO 6.3.2.7-10 2.4.1.69 O 1.2.3.7 2.4.2.19 R OPPT OH OH + 1.1.1.22 UDPglucose dehydrogenase 2.4.99.7 HO O OPPU HO 2.7.1.32 Choline kinase S CH2OH 6.3.2.13 OH OPPU CH HO CH2CH(NH3)COO HO CH CH NH HO CH2CH2NHCOCH3 CH O CH CH NHCOCH COO 1.1.1.23 Histidinol dehydrogenase OPC 2.4.1.17 3 2.4.1.29 CH CHO 2 2 2 3 2 2 3 O 2.7.1.33 Pantothenate kinase CH3CH NHAC OH OH OH LACTOSE 2 COO 1.1.1.25 Shikimate dehydrogenase A HO HO OPPG CH OH 2.7.1.34 Pantetheine kinase UDP- TDP-Rhamnose 2 NH NH NH NH N M 2.7.1.36 Mevalonate kinase 1.1.1.27 Lactate dehydrogenase HO COO- GDP- 2.4.1.21 O NH NH 4.1.1.28 2.3.1.5 2.1.1.4 1.1.1.29 Glycerate dehydrogenase C UDP-N-Ac-Muramate Iduronate OH 2.4.1.1 2.4.1.11 HO 5-Hydroxy- 5-Hydroxytryptamine N-Acetyl-serotonin N-Acetyl-5-O-methyl-serotonin Quinolinate 2.7.1.39 Homoserine kinase Mannuronate CH3 etc.